Purification of recombinant human RAD52
Human RAD52 cDNA was codon optimized for expression in E. coli and cloned into pET100 (GeneArt, Thermo Fisher Scientific). Inverse PCR (primers: RAD52_tag_remove_F and RAD52_tag_remove_R) was performed to remove the 6×His, T7 and Xpress tags. The plasmid was transformed into BL21 Star (DE3) (Thermo Fisher Scientific) cells and a single colony was inoculated into an overnight culture using Luria broth (LB) supplemented with 0.8% glucose and 100 μg ml−1 ampicillin. An aliquot was diluted into 2 l of LB containing glucose and ampicillin, to an optical density at 600 nm (OD600) of 0.1, and incubated in an orbital shaker at 37 °C and 180 rpm. When the culture reached an OD600 of 0.8, IPTG (0.5 mM; Thermo Fisher Scientific) was added to induce RAD52 expression and incubation continued for a further 3 h. The culture was collected by centrifugation at 3,300g for 15 min, and the cell pellet was resuspended in 1 vol of PBS and centrifuged again. The pellet was then resuspended in lysis buffer (25 mM MES pH 6.5, 0.5 M NaCl, 10% glycerol and 1 mM EDTA) supplemented with Halt protease inhibitor (Thermo Fisher Scientific) and 0.25 mM TCEP, and lysed with Emulsiflex C5 (Avestin) at 4 °C. The lysate was clarified by centrifugation at 60,000g and 4 °C for 10 min. The supernatant was collected and diluted dropwise with the same lysis buffer without NaCl to reach 300 mM NaCl. The lysate was then clarified again by centrifugation at 60,000g and 4 °C for 20 min and loaded onto a HiTrap SP column (Cytiva) connected to an ÄKTA pure system at 4 °C. The column was washed with 3 column volumes (CV) of buffer containing 25 mM MES pH 6.5, 0.3 M NaCl, 1 mM EDTA, 10% glycerol and 0.25 mM TCEP, and eluted with 10 CV of a linear gradient of the same buffer containing 0.3–1 M NaCl. Peak fractions were diluted 3× with buffer containing 100 mM HEPES pH 7.0, 0.25 mM TCEP and Halt protease inhibitor and loaded onto a HiTrap Q column (Cytiva), that was eluted with 10 CV of a linear gradient (0.1–1 M NaCl) of HEPES buffer containing 0.5 mM EDTA and 0.25 mM TCEP. The HiTrap Q flow-through fraction was collected as crude purified RAD52.
To separate the two RAD52 conformations, RAD52 was loaded onto a Resource S column (Cytiva). Chromatography was performed using buffer containing 25 mM HEPES pH 7.0, 0.25 mM TCEP and various concentrations of NaCl. The Resource S column was (1) washed with 3 CV of 150 mM NaCl buffer; (2) eluted with 5 CV of linear gradient of 0.2–0.278 M NaCl buffer (until the conductivity was equivalent to 24.4 mS cm−1); (3) washed with 5 CV of 0.278 M NaCl buffer; and (4) eluted with 10 CV of 0.278–0.6 M NaCl buffer. The peak fractions of the two RAD52 forms were collected separately. RAD52-OR and RAD52-CR were loaded onto a Superose 6 Increase 10/300 GL column (Cytiva) using buffer containing 25 mM HEPES pH 8.0, 200 mM KOAc, 10% glycerol and 0.25 mM TCEP. The peak fractions were collected, aliquoted, snap-frozen in liquid nitrogen and stored at −80 °C. RAD52 concentrations were measured at a wavelength of 280 nm using the Nanodrop (Thermo Fisher Scientific) system and calculated as an 11-subunit ring (RAD52-CR and RAD52 NTD) or 10-subunit ring (RAD52-OR) with the exception that protomer concentration was used for circular dichroism (CD) analyses.
For the RAD52 NTD, inverse PCR (primers: RAD52_NTD_F and RAD52_NTD_R) was used to remove the C terminus (amino acids 210–418). The RAD52 NTD was purified using the same method as the full-length protein except that a linear gradient of 0.2–0.6 M NaCl was used for the Resource S column.
For RAD52(∆RID), RAD52(RQK/AAA) and RAD52(∆C), inverse PCR was used to remove the RPA-interacting domain (primers: RAD52_RID_F and RAD52_RID_R), extreme C terminus (primer: RAD52_NTD_F and RAD52_C_18D_R) and introduce the R260A, Q261A and K262A mutations (primer: RAD52_RQKAAA_F and RAD52_RQKAAA_R). All mutants were purified using the same method as for the full-length protein.
Purification of Flag–RAD52 from Sf9 insect cells
Human RAD52 cDNA was codon optimized for expression in Sf9 insect cells and cloned into pFastBac1 baculovirus expression vector with an N-terminal Flag tag (GeneArt, Thermo Fisher Scientific). The plasmid was transformed into DH10Bac (Thermo Fisher Scientific), and the bacmids were isolated with PureLink HiPure Plasmid Miniprep kit (Thermo Fisher Scientific). Overall, the generation and handling of the baculovirus was performed according to the Invitrogen Bac-to-Bac Baculovirus Expression System user manual with some modifications. In brief, recombinant bacmids were transfected into Sf9 cells with FuGENE HD, and P1 viruses were collected 66–72 h after transfection. The baculovirus titre was determined by isolating the viral DNA with High Pure Viral Nucleic Acid Kit (Roche), and quantitative PCR using Platinum qPCR supermix UDG (Thermo Fisher Scientific) and BaculoQUANT kit (Oxford Expression Technologies). The P2 baculovirus was amplified by infecting Sf9 cells at a multiplicity of infection (MOI) of 0.01 and 2 million cells per ml, and collected at 66–72 h after infection.
P2 baculovirus (MOI = 1) was used for recombinant Flag–RAD52 expression. Sf9 cells were grown in Sf-900 III SFM (Gibco, Thermo Fisher Scientific) at 27 °C in an orbital shaker at 140 rpm. The Sf9 cells were infected for 66–72 h. Cells were collected by centrifugation at 500g for 5 min and washed once with PBS. The cell pellet was resuspended in lysis buffer (25 mM MES pH 6.5, 600 mM NaCl, 10% glycerol and 1 mM EDTA) supplemented with Halt protease inhibitor (Thermo Fisher Scientific) and 0.25 mM TCEP, and sonicated in ice/water slurry at 25 amplitude for 150 s (with 1 s intervals to prevent warming) using a qSonica Q700 sonicator. The lysate was clarified by centrifugation at 60,000g for 30 min at 4 °C.
Pre-equilibrated anti-Flag M2 agarose beads (Merck) were added to the lysate, and the mixture was incubated on a rotator at 4 °C for 1.5 h. The beads were pelleted by centrifugation at 500g for 5 min at 4 °C and transferred to a gravity flow chromatography column. The column was washed extensively with the lysis buffer, and subsequently with buffer containing 25 mM HEPES pH 7.0, 450 mM NaCl, 10% glycerol, 1 mM EDTA, 0.25 mM TCEP and Halt protease inhibitor. The last wash was performed with the same buffer at 300 mM NaCl. Flag–RAD52 was then eluted with the buffer containing 450 mM NaCl and 0.5 mg ml−1 Flag peptide. Elution was performed twice by incubating the beads with an equal volume of elution buffer for 1 h at 4 °C. The eluates were combined, and diluted 4× using the same elution buffer at 100 mM NaCl without Flag peptide to lower the NaCl concentration to 150 mM. Resource S chromatography was performed as described above.
Purification of recombinant human RPA
Human RPA1, RPA2 and 10×His-RPA3 were synthesized and cloned into the pFastBac1 baculovirus expression vector (GeneArt, Thermo Fisher Scientific). RPA1 (2 copies), RPA2 and 10×His-RPA3, together with their polyhedrin promoters, were then assembled into pBIG1a (biGBac multigene baculovirus expression vector)49 using Gibson assembly (NEB). Bacmids and baculovirus were generated as described above. P2 baculovirus (MOI = 1) was used for recombinant RPA expression. Sf9 cells were grown in Sf-900 III SFM (Gibco, Thermo Fisher Scientific) at 27 °C in an orbital shaker at 140 rpm, and infected for 66–72 h. Cells were collected by centrifugation at 500g for 5 min and washed once with PBS. The cell pellet was resuspended in buffer containing 25 mM HEPES pH 8.0, 0.5 M NaCl, 10% glycerol, 0.01% Tween-20, 20 mM imidazole, Halt protease inhibitor and 0.25 mM TCEP, and sonicated in ice/water slurry at 25 amplitude for 150 s (with 1 s interval to prevent warming) with a qSonica Q700 sonicator. The lysate was clarified by centrifugation at 60,000g for 30 min at 4 °C.
Pre-equilibrated Ni-NTA beads (Qiagen) were added to the lysate and the mixture was incubated on a rotator at 4 °C for 1 h. The beads were pelleted by centrifugation at 500g for 5 min at 4 °C and transferred to a chromatography column. The column was washed extensively with lysis buffer excluding Tween-20 while gradually decreasing the NaCl concentration from 0.5 to 0.2 M. Recombinant RPA was eluted with buffer containing 25 mM Tris-HCl pH 8.0, 0.2 M NaCl, 10% glycerol, 250 mM imidazole, Halt protease inhibitor and 0.25 mM TCEP. The RPA eluate was diluted 2× with the same elution buffer, without NaCl and imidazole, to lower the NaCl concentration to 100 mM. The diluted eluate was then loaded onto a Resource Q column (Cytiva) and eluted with linear gradient of buffer containing 0.1–0.4 M NaCl, 25 mM HEPES pH 8.0, 10% glycerol and 0.25 mM TCEP. Peak fractions containing RPA were loaded onto a Superdex 200 Increase 10/300 GL column (Cytiva) using buffer containing 25 mM HEPES pH 8.0, 200 mM KOAc, 0.5 mM EDTA, 10% glycerol and 0.25 mM TCEP. The protein was collected, aliquoted, snap-frozen in liquid nitrogen and stored at −80 °C.
For RPA1(∆FAB) and RPA2(∆WHD), inverse PCR was used to remove the DBD-F, DBD-A and DBD-B of RPA1 (amino acids 2–440) (primers: DBDC_F and DBDC_R) and the WHD of RPA2 (amino acids 207–270) (primers: RPA2_WHD_F and RPA2_WHD_R). Both deletion mutants were purified using the same method as described for the full-length protein.
Oligonucleotides
All DNA oligonucleotides were HPLC purified (Merck and Integrated DNA Technologies). The names and sequences of the oligos were as follows where FAM is 6-carboxyfluoroscein: RAD52_tag_remove_F (5′-AGCGGCACCGAAGAAGCAATTTTAGG-3′), RAD52_tag_remove_R (5′-CATATGTATATCTCCTTCTTAAAGTTAAACAAAATTATTTCTAGAGGGG-3′), RAD52_NTD_F (5′-TAAAAGGGCGAGCTCAACGATCCGGCTG-3′), RAD52_NTD_R (5′-ACGACAGCTATTATAACGTGCTTCTTCAACGCTCGG-3′), RAD52_RID_F (5′-CCTCCGGCACCGCCTGTTAC-3′), RAD52_RID_R (5′-ATCCTGATCTGCCGGAATAACTGCATG-3′), RAD52_RQKAAA_F (5′-CGCACAGCTGCAACAGCAGTTTCGTGAACGTATGG-3′), RAD52_RQKAAA_R (5′-GCAGCCAGTTTACGCTGATGGGTTGCTTCGCTTTCAACTGCG-3′), RAD52_C_18D_R (5′-ATTACCGGTGGTACGCTGATCTGCGCTATAGG-3′), DBDC_F (5′-AACTGGAAAACCTTGTATGAGGTCAAATCCGAGAACCTGGG-3′), DBDC_R (5′-CATGGATCCGCGCCCGATGGTGG-3′), RPA2_WHD_F (5′-GCGGCCGCTTTCGAATCTAGAGCCTG-3′), RPA2_WHD_R (5′-AGTGAGGCCATTTGCTGGCATGAAGCTATTCC-3′), SSA1 (5′-TATCGAATCCGTCTAGTCAACGCTGCCGAATTCTACAGAGTTTGGGCTCCTCAACCTGCAGGTT-3′), SSA2 (5′-AACCTGCAGGTTGAGGAGCCCAAACCTCACTGGTAAATTCGCAGCGTTGACTAGACGGATTCGATA-3′), FAM-SSA4 (40nt) (5′-FAM-TATCGAATCCGTCTAGTCAACGCTGCCGAATTCTACCAGT-3′), SSA5 (5′-ACTGGTAGAATTCGGCAGCGTTGACTAGACGGATTCGATA-3′), SSA6 (5′-TGACCATCTTAAGCCGTCGCAACTGATCTGCCTAAGCTAT-3′), SSA7 (5′-CGGCAGCGTTGACTAGACGGATTCGATA-3′), gap 1-1 (5′-CGTGAAGTCGCCGACTGAATGCCAGCAATCTCTTTTTGAGTCTCATTTTGCATCTCGGCAATCTCTTTCTGATTGTCCAGTTGCATTTTAGTAAGCTCTTTTTGATTCTCAAATCCGGCG-3′), gap 1-2 (5′-CGCCGGATTTGAGAATCAAAAAGAGCTTAC-3′) and gap 1-3 (5′-GATTGCTGGCATTCAGTCGGCGACTTCACG-3′). Cy3- and Cy5-labelled and biotinylated oligonucleotides were purchased (Merck). To generate FAM-SSA1/SSA2 dsDNA, equimolar concentrations of FAM-SSA1 and SSA2 were mixed in 10 mM Tris-HCl pH 7.5, 100 mM NaCl and 1 mM EDTA, heated to 90 °C and gradually cooled to room temperature. Gapped DNA was annealed as described using gap 1-1, gap 1-2 and gap 1-3. Concentrations were measured using a spectrophotometer using absorbance values at 260 nm. All DNAs were stored at −20 °C.
Fluorescence anisotropy
DNA-binding reactions (20 μl) were performed at 25 °C in buffer containing 25 mM HEPES pH 8.0, 0.2 M KOAc, 10% glycerol, 0.25 mM TCEP, 1 mM Mg(OAc)2 and 0.01% Brij-35. Proteins were serially diluted and mixed with 10 nM (final concentration) of FAM-labelled DNA in 384-well microplates (Corning). The plates were measured using the CLARIOstar microplate reader (BMG Labtech). Blank-corrected anisotropy measurements were averaged and plotted against protein concentration. RAD52 binding was curve-fitted using the following quadratic equation in GraphPad Prism 9 to determine KD values:
$$Y={A}_{\min }+\left({A}_{\max }{-A}_{\min }\right)\times \frac{x+L+{K}_{{\rm{D}}}-\sqrt{{\left(x+L+{K}_{{\rm{D}}}\right)}^{2}-4\times x\times L}\,}{2\times L},$$
where Y is the fluorescence anisotropy, Amin and Amax are the minimum and maximum fluorescence anisotropy values, L is the ligand concentration (equal to 0.01 µM), x is the protein concentration and KD is the dissociation constant. At least three independent triplicates of technical replicates were performed for each binding condition.
Single-stranded DNA annealing
Reactions (15 μl) contained 5′-32P-labelled SSA1 (68 nucleotides) with its complementary strand SSA2 (68 nucleotides)30 in 25 mM HEPES pH 8.0, 0.2 M KOAc, 1 mM Mg(OAc)2, 0.01% Brij-35, 0.25 mM TCEP and 5% glycerol. Two separate 7.5 µl reaction mixtures were set up. One contained 5′-32P-labelled SSA1 (0.33 nM) in buffer, and the second contained SSA2 (0.33 nM). RPA (0.33 nM) was added to both, as indicated. RAD52 (0.33 nM, or as indicated in figure legends) was added to SSA2 and incubated for 10 min at 25 °C. The two tubes were then mixed and incubated for 10 min at 25 °C, before being stopped by deproteinization using 3 µl of proteinase K (20 mg ml−1 proteinase K in 10 mM Tris-HCl pH 7.5 and 1 mM CaCl2) and incubated at 30 °C for 30 min. The samples were supplemented with Ficoll loading buffer and analysed by PAGE with TBE as the running buffer. Gels were dried and exposed to phosphorimaging plates and images acquired using the Typhoon FLA 9500 biomolecular imager (GE) and quantified using ImageJ50,51.
For reactions using 40-nucleotide ssDNA (5′-32P-labelled SSA4 with complimentary SSA5), the reactions were set up as described above except that the concentration of ssDNA was lowered to 0.13 nM to prevent self-annealing of ssDNA, and 0.13 nM of RPA was used. Concentrations of RAD52 are indicated in figure legends.
To determine whether DNA ends were required for RAD52-OR mediated annealing, interactions between 0.33 nM circular φX174 virion ssDNA and 0.33 nM 32P-labelled gapped duplex DNA (a 60-nucleotide-long ssDNA that had 30-mers annealed to each end) were analysed. For these experiments, RPA (0.33 nM or 19.9 nM) was premixed with the gapped and circular ssDNAs, respectively (to provide similar coverage). RAD52 was then added to the gapped ssDNA and annealing was measured by electrophoresis through a 1% agarose gel using TAE buffer.
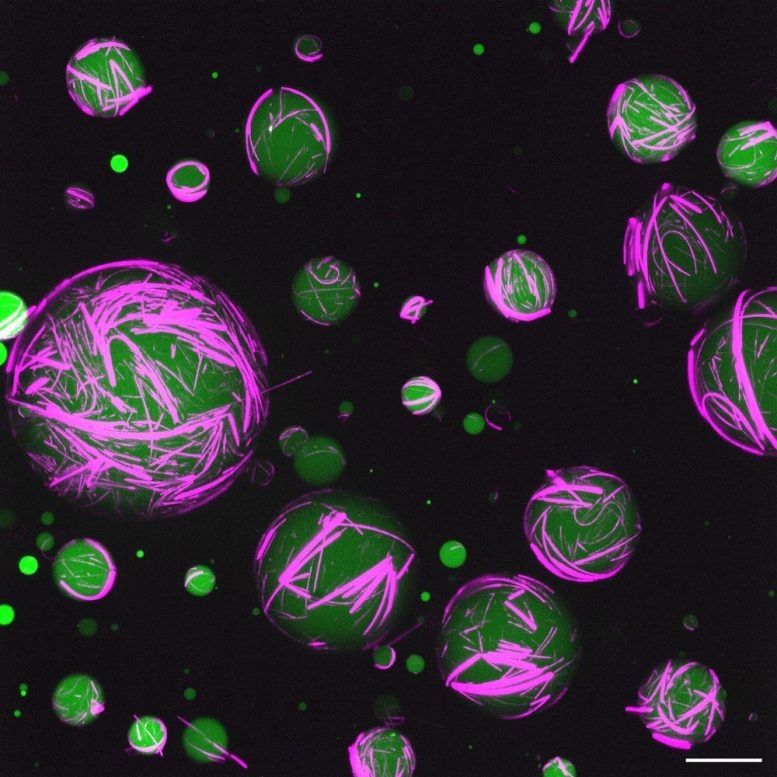
To analyse ssDNA annealing using size-exclusion chromatography, RAD52-OR (4 µM) was preloaded on SSA2–Cy5 (4 µM, 12.5 µl) before an equal volume of Cy3–SSA1 (4 µM) was added. After 30 min on ice, the reaction was loaded onto the Superdex 200 Increase 3.2/300 column connected to the ÄKTA pure Micro system. Chromatography was performed at 4 °C with buffer containing 25 mM HEPES pH 8.0, 200 mM KOAc, 0.25 mM TCEP and 1 mM Mg(OAc)2.
Biolayer interferometry analysis
40-nucleotide (SSA4) ssDNA was biotinylated at either the 5′ or 3′ end (indicated as bio–ssDNA or ssDNA–bio, respectively). 68-nucleotide (SSA1) ssDNA was biotinylated at the 3′ end (indicated as SSA1–bio), and 28 nucleotides of complementary ssDNA was annealed to the 5′ end to protect the 5′ ssDNA end (indicated as ds-ssDNA–bio). The experiments were performed using the Octet R8 system (Sartorius) at 25 °C in buffer containing 25 mM HEPES pH 8.0, 200 mM KOAc, 0.01% Tween-20, 1 mM Mg(OAc)2 and 0.25 mM TCEP. The biotinylated DNA substrates (5 nM) were immobilized onto Octet SA streptavidin biosensors until a 0.05 threshold, and the sensors were then moved to wells containing a range of RAD52 concentrations (20, 10, 5, 2.5, 1.25, 0.625 and 0.312 nM). The association of RAD52 to DNA was recorded for 60 min and the dissociation for 5 min using the Octet BLI Discovery Software. Equilibrium dissociation constants (KD) were obtained by plotting association amplitudes at equilibrium versus protein concentration (Octet Analysis Studio Software; Sartorius) and plotted in GraphPad Prism 9. The following 1:1 binding equation was used to determine KD values: using the following quadratic equation in GraphPad Prism 9 to determine KD values:
$$Y={B}_{\max }\times X/({K}_{D}+X),$$
where Y is the association amplitude, Bmax is the maximum amplitude at saturation, X is the protein concentration and KD is the dissociation constant. Three independent triplicates were performed for each binding condition.
CD analysis
Far-UV CD measurements were performed on a Jasco J-815 spectropolarimeter fitted with a cell holder temperature-regulated by a CDF-426S Peltier unit. Spectra were recorded at 20 °C at protein concentrations of 3.3 µM (RAD52-OR) and 3.2 µM (RAD52-CR) in 10 mM potassium phosphate buffer pH 8.0, 100 mM NaF and 0.25 mM TCEP. Fused silica cuvettes were used with a 1 mm path length (Hellma). Spectra were recorded at a resolution of 0.2 nm and were baseline corrected by subtraction of the appropriate buffer spectrum. CD intensities are presented as the molar CD extinction coefficient (∆εM) calculated as:
$${\Delta \varepsilon }_{{\rm{M}}}=\frac{S}{\mathrm{32,980}\times {c}_{{\rm{M}}}\times L}\left({\rm{units:}}{{\rm{M}}}^{-1}{{\rm{cm}}}^{-1}\right),$$
where S is the signal in millidegrees, cM is the molar concentration and L is the path length (in cm). Secondary structure content was estimated as described52.
Intact protein MS
Proteins were diluted to 1 µM with 0.1% (v/v) formic acid and injected onto a C4 BEH 1.7 µm, 1.0 × 100 mm, UPLC column using the Acquity I class LC (Waters) system. Proteins were eluted with a 15 min gradient of acetonitrile (2% (v/v) to 80% (v/v)) in 0.1% (v/v) formic acid using a flow rate of 50 µl min−1. The analytical column outlet was directly interfaced through an electrospray ionization source, with a time-of-flight (TOF) mass spectrometer (BioAccord, Waters). Data were acquired over a m/z range of 300–8,000, in positive-ion mode with a cone voltage of 40 V. Scans were summed together manually and deconvoluted using MaxEnt1 (Masslynx, Waters). The parameters used were as follows; input m/z range (Da): 600–2,000; output mass range (Da): 30000–60000; TOF resolution: 10000.00; and iterate to convergence.
GuHCl denaturation and renaturation
RAD52 (purified to the HiTrap Q step) was dialysed into 25 mM HEPES pH 7.0, 6 M GuHCl, 0.5 mM EDTA and 2 mM β-mercaptoethanol overnight at 4 °C. The denatured protein was analysed using a Superose 6 Increase 10/300 GL column, which was run with 6 M GuHCl buffer. Protein was renatured by dialysis in native buffer (25 mM HEPES pH 7.0, 200 mM NaCl, 0.5 mM EDTA and 2 mM mercaptoethanol) for 24 h at 4 °C. The renatured RAD52 was then run on the same column using native buffer. To analyse the percentage of open and closed rings, the renatured RAD52 sample was loaded onto the Resource S column.
Negative-stain EM sample preparation and data acquisition
Samples (4 µl, 25 ng µl−1) were applied for 1 min to glow discharged (25 mA, 30 s) 400-mesh carbon-coated copper grids (C400Cu100, EM Resolutions). The grids were sequentially stained in four separate 30 µl droplets of 2% (v/v) uranyl acetate for 10, 15, 20 and 25 s. Excess uranyl acetate was blotted away from the grid using Whatmann paper, allowed to air dry and stored before imaging.
The grids were imaged on the Tecnai LaB6 G2 Spirit TEM operating at 120 kV equipped with a 2K Gatan Ultrascan 1000 camera. Micrographs were acquired manually using DigitalMicrograph at a nominal magnification of ×30,000 (3.5 Å per pixel) or ×42,000 (2.4 Å per pixel) with defocus values ranging from −0.7 to −1.5 µm.
Negative-stain EM data analysis
DM3 files were converted to MRC format using e2proc2d.py (EMAN2)53. Micrographs were imported into Relion 3.1 or 4.154,55, CTF parameters were calculated using CTFFIND456, and particles were picked using crYOLO57 or Topaz58. Particles were extracted and iteratively 2D classified (ignore CTF to first peak = yes, limit resolution E-step = 20 Å, additional arguments = –only-flip-phases).
Cryo-EM sample preparation
Recombinant RAD52 and RPA were purified to the Resource S or Resource Q step, and freshly purified on the Superose 6 Increase 10/300 GL or Superdex 200 Increase 10/300 GL column before making the cryo-EM grids. For RAD52-CR, the protein was in a buffer containing 25 mM HEPES pH 7.0, 150 mM NaCl and 0.25 mM TCEP, diluted to 0.3 mg ml−1, and supplemented with 0.00005% Tween-20. A sample (4 μl) was applied to freshly glow-discharged (45 mA, 60 s; Quorum Emitech K100X) Quantifoil R2/1 300 mesh copper grids and vitrified using a Vitrobot Mark IV (Thermo Fisher Scientific) cooled to 4 °C with 95% humidity. Grids were double-side blotted for 0.5 s and plunge frozen in liquid ethane. For RAD52-OR, the grids were prepared as described above except Quantifoil R2/2 200 mesh copper grids were used, and the concentration was 0.25 mg ml−1, the Tween-20 concentration was 0.001%, and blot time was 1.5 s. For RAD52-OR–ssDNA, the protein (0.25 mM) was diluted to 0.5 µM in 25 mM HEPES pH 8.0, 150 mM NaCl, 2 mM Mg(OAc)2 and supplemented with 0.05% octyl-β-glucoside (OG). SSA4 (1 µM) was added and incubated at 25 °C for 10 min. The concentration was determined by Bradford assay (Bio-Rad) and diluted to 0.15 mg ml−1 with the same buffer. Grids were prepared as above except Quantifoil R1.2/1.3 300 mesh copper grids were used and the blot time was 2.5 s. For RPA–ssDNA, the protein (0.25 mM), in 25 mM HEPES pH 8.0, 150 mM NaCl, 2 mM Mg(OAc)2, was diluted to 3 µM, and supplemented with 0.1 mM CHAPSO. SSA7 (6 µM) was added and incubated at 25 °C for 10 min. The concentration was determined using the Bradford assay (Bio-Rad) and diluted to 0.15 mg ml−1 with the same buffer. UltrAuFoil R2/2 200 mesh gold grids (Quantifoil) were prepared as described above and the blot time was 2.5 s. The RAD52-OR–ssDNA–RPA ternary complex was assembled as indicated in the ‘Reconstitution of the RAD52–ssDNA–RPA complex’ section below. The concentration was determined using the Bradford assay (Bio-Rad) and diluted to 0.1 mg ml−1 with buffer supplemented with 0.00075% Tween-20 and 0.075 mM CHAPSO. Quantifoil R2/2 200 mesh copper grids were prepared as described above, except the blot time was 3 s.
Cryo-EM data collection, image processing and atomic model building
RAD52-CR and RAD52-OR datasets were collected on a Titan Krios Cryo-TEM equipped with a Falcon III direct electron detector (Thermo Fisher Scientific) at the Francis Crick Institute Structural Biology STP. The RAD52-OR–ssDNA dataset was collected on a Titan Krios G3i (FEI, Thermo Fisher Scientific) equipped with a Gatan K3 direct electron detector at the London consortium for cryo-EM (LonCEM). RPA–ssDNA and RAD52-OR–ssDNA–RPA datasets were collected on a Titan Krios Cryo-TEM (Thermo Fisher Scientific) equipped with a K2 direct electron detector (Gatan) at the Francis Crick Institute Structural Biology STP.
Single-particle analyses were performed within Relion (v.4.0)54 and CryoSPARC59. The videos were corrected for drift and dose-weighted using RELION’s own implementation of MOTIONCOR260 and subsequent contrast transfer (CTF) parameters were measured using CTFFIND456. Particles were picked automatically using crYOLO57 or Topaz58. Details of image processing are illustrated in Extended Data Figs. 3, 4, 5, 8 and 9. In brief, several rounds of 2D classification were performed to remove particles that cannot be aligned to yield defined 2D averages. Several rounds of 3D classifications were performed to separate different conformations or particles that cannot be aligned to yield high-resolution 3D volumes. 3D auto-refine, Bayesian polishing (minimum two rounds) and CTF refinement (minimum one round) were performed iteratively to achieve high resolution 3D reconstruction in RELION61,62. Polished particles were imported to CryoSPARC59, and refined using non-uniform refinement63. 3D variability64 or 3D classifications were performed to detect heterogeneity within the cryo-EM densities. The cryo-EM maps were sharpened by post-processing in RELION, CryoSPARC or DeepEMhancer65 if there was high variability in local resolution. The overall resolution is reported at FSC = 0.143 (ref. 66).
All model building was performed using Phenix67,68, COOT69 and ISOLDE70 in ChimeraX71. For RAD52-CR, the crystal structure of the RAD52 NTD (PDB: 1H2I) was placed into a sharpened RAD52-CR cryo-EM map in ChimeraX71 and initially refined using Namdinator72. One RAD52 subunit was removed from RAD52-CR and used for initial refinement in Namdinator for RAD52-OR. ssDNA was built manually in COOT into the RAD52-OR model using RAD52-OR–ssDNA as a starting model. RPA1, RPA2 and RPA3 AlphaFold2 models were used for Dock and rebuild in Phenix73,74 and the ssDNA model was aligned and extracted from the fungal RPA structure (PDB: 4GOP)39. The RAD52-OR–ssDNA model was used as the initial model for RAD52-OR–ssDNA–RPA.
SEC–MALLS analysis
SEC–MALLS was used to determine the molar mass composition of RAD52. Purified RAD52-OR (2.0, 1.0 or 0.5 mg ml−1) was loaded onto a Superose 6 Increase 10/300 GL column connected to a Jasco chromatography system. Chromatography was performed at 25 °C with buffer containing 25 mM HEPES pH 7.0, 150 mM NaCl, 0.25 mM TCEP and 3 mM NaN3 at a flow rate of 1.0 ml min−1. RAD52-OR–ssDNA (2 mg ml−1) was analysed in a similar manner using 25 mM Bis-Tris propane pH 8.5, 200 mM NaCl, 5 mM MgCl2, 0.25 mM TCEP and 3 mM NaN3 as the running buffer. The scattered light intensity and protein concentrations of the column eluates were recorded using a DAWN-HELEOS laser photometer and an OPTILAB-rEX differential refractometer (dn/dc = 0.186). The weight-averaged molecular mass of material contained in chromatographic peaks was determined using the combined data from both detectors in the ASTRA software v.7.3.2 (Wyatt Technology).
Nuclear/chromatin extraction and analysis
U2OS cells (authenticated and microplasma free, as determined by the Francis Crick Institute) were grown in DMEM (Gibco) supplemented with 10% FBS (Gibco) in humidified incubators at 37 °C and 5% CO2. Cells were collected from four confluent 500 cm2 square dishes and washed once with PBS. The pellet was supplemented with 5× pellet volume of CSK buffer (10 mM PIPES pH 6.8, 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 1 mM EGTA, 0.5% Triton X-100 and 0.25 mM TCEP) supplemented with Halt protease and phosphatase inhibitors, incubated on ice for 10 min and centrifuged at 2,000g for 5 min at 4 °C. The supernatant was collected as the first CSK extract. A 3× pellet volume of CSK buffer (containing 0.1% Triton X-100) was added to the pellet, incubated on ice for 10 min and the sample was centrifuged at 3,000g for 5 min at 4 °C. The supernatant was collected as the second CSK extract. An equal volume of benzonase digestion buffer (20 mM HEPES pH 8.0, 2 mM MgCl2, 0.5% Triton X-100, 0.25 mM TCEP and 500 units benzonase/100 µl of buffer) supplemented with Halt protease and phosphatase inhibitors was added to the pellet and incubated on ice for 10 min. A 2× sample volume of high-salt buffer (20 mM HEPES pH 8.0, 600 mM NaCl and 0.25 mM TCEP) supplemented with Halt protease and phosphatase inhibitors was then added, incubated on ice for 10 min, and the sample was centrifuged at 21,000g for 10 min at 4 °C. The supernatant was collected as a nuclear/chromatin extract.
Glycerol gradients (5 ml, 10–30%) in 25 mM HEPES pH 8.0, 150 mM NaCl, 10–30% glycerol and 0.25 mM TCEP were cast in thin-wall polypropylene tubes (Beckman Coulter) using a Gradient Master (Biocomp) and kept in the cold room overnight to equilibrate to 4 °C. U2OS nuclear/chromatin extracts (200 µl), 200 ng recombinant RAD52-OR or a gel-filtration calibration marker (Cytiva) was loaded gently onto the top of three gradients, which were then centrifuged at 4 °C and 55,000 rpm (368,000g) using SW 55 Ti rotor (Beckman Coulter) for 4 h. The fractions were collected by manual pipetting from the top of the gradients. The U2OS nuclear/chromatin extract (500 µl), 500 ng recombinant RAD52-OR or a gel-filtration calibration marker (Cytiva) were also loaded onto the pre-equilibrated Superose 6 Increase 10/300 GL column (Cytiva). Chromatography was performed with a buffer containing 25 mM HEPES pH 8.0, 150 mM NaCl, 10% glycerol and 0.25 mM TCEP at 4 °C. Fractions were collected and analysed by SDS–PAGE followed by western blotting using antibodies against RAD52 (rabbit monoclonal, 1:500, Abcam, ab124971). Alexa Fluor Plus 800 anti-rabbit secondary antibodies (1:2,000, Invitrogen, A32735) were used and the membranes were imaged using an Odyssey DLx instrument with ImageStudio software (Licor).
RAD52 Resource S chromatogram peak fitting
Resource S chromatography was performed as described above except a linear gradient of 0.2–0.6 M NaCl was used. The UV280 absorbance values were imported into GraphPad Prism 9 and curved fitted using a sum of two Gaussians equation to deconvolute open- and closed-ring peaks:
$$Y={\rm{amplitude}}\times \exp \left(-0.5\times {\left(\frac{X-{\rm{mean}}}{{\rm{s.d.}}}\right)}^{2}\right)+\mathrm{amplitude\; 2}\times \exp \left(-0.5{\left(\frac{X-\mathrm{mean\; 2}}{\mathrm{s.d.\; 2}}\right)}^{2}\right)$$
RAD52–ssDNA–RPA pull downs
The RAD52–ssDNA–RPA ternary complex (400 μl) was reconstituted in buffer containing 25 mM HEPES pH 8.0, 200 mM KOAc, 2 mM Mg(OAc)2, 0.01% Tween-20 and 0.25 mM TCEP. Biotin-labelled SSA4 (0.1 μM), with photo-cleavable linker (Integrated DNA Technologies), and recombinant RPA (0.15 μM) were mixed and incubated on ice for 10 min. RAD52-OR (0.15 μM) was then added and incubation continued for a further 10 min. Pre-washed Streptavidin Sepharose Mag beads (10 μl, Cytiva) were then added and incubated for 30 min on a head-to-toe rotator at 4 °C. The beads were washed once with reaction buffer and then with reaction buffer Tween-20. The beads were resuspended in 20 μl reaction buffer, and irradiated with 365 nm UVA on ice/water slurry to cleave the photo-cleavable linker.
Reconstitution of the RAD52–ssDNA–RPA complex
RAD52-OR (purified to the Resource S step) and RPA (purified to the Resource Q step) were loaded onto the Superose 6 Increase 10/300 GL (Cytiva) and Superdex 200 Increase 10/300 GL (Cytiva) columns, respectively, and run with buffer containing 25 mM HEPES pH 8.0, 150 mM NaCl, 2 mM Mg(OAc)2 and 0.25 mM TCEP. The reconstitution mixture for cryo-EM was supplemented with 0.00075% Tween-20 and 0.075 mM CHAPSO, whereas the XL-MS sample was supplemented with 0.05% OG. Reconstitution of the RAD52-OR–ssDNA–RPA ternary complex involved two steps: (1) RPA (1 µM final concentration) was added to SSA1 (0.5 µM final concentration) and incubated at 25 °C for 10 min; and (2) RAD52-OR (0.5 µM final concentration) was added and incubated at 25 °C for 30 min. The sample was centrifugated at 21,000g for 1 min at 4 °C before proceeding with cryo-EM grid preparation and XL-MS.
Protein disorder prediction
The human RAD52 protein sequence (UniProt: P43351) was uploaded to the ODiNPred75 webserver (https://st-protein.chem.au.dk/odinpred). The predicted disorder probability of each residue was plotted in GraphPad Prism 9.
Multiple-sequence alignment
RAD52 protein sequences from different organisms were aligned with Clustal Omega using the default settings76. The alignment was formatted with ESPript3.077.
XL-MS analysis
RAD52-OR and RAD52-OR–ssDNA–RPA ternary complexes (0.5 µM, reconstituted as above) were supplemented with a 1:100 molar ratio of disuccinimidyl dibutyric urea (DSBU: 50 µM) for 1 h at room temperature, before the mixture was quenched by the addition of NH4HCO3 to a final concentration of 20 mM (15 min at room temperature). The cross-linked proteins were reduced with 10 mM dithiothreitol and alkylated with 50 mM iodoacetamide. They were then digested with trypsin at an enzyme-to-substrate ratio of 1:100, for 1 h at room temperature and further digested overnight at 37 °C after addition of trypsin at a ratio of 1:20. The peptide digests were then fractionated batch-wise by high pH reverse-phase chromatography on micro spin TARGA C18 columns (Nest Group) into four fractions (10 mM NH4HCO3/10% (v/v) acetonitrile pH 8.0; 10 mM NH4HCO3/20% (v/v) acetonitrile pH 8.0; 10 mM NH4HCO3/40% (v/v) acetonitrile pH 8.0; and 10 mM NH4HCO3/80% (v/v) acetonitrile pH 8.0). The fractions (150 µl) were evaporated to dryness in a CentriVap concentrator (Labconco) before analysis by LC–MS/MS.
Lyophilized peptides were resuspended in 1% (v/v) formic acid and 2% (v/v) acetonitrile and analysed by nano-scale capillary LC-MS/MS using a Vanquish Neo UPLC (Thermo Fisher Scientific, Dionex) to deliver a flow of approximately 300 nl min−1. A PepMap Neo C18 5 μm, 300 μm × 5 mm nanoViper (Thermo Fisher Scientific, Dionex) trapped the peptides before separation on a 25 cm EASY‐Spray column (25 cm × 75 µm inner diameter, PepMap C18, 2 µm particles, 100 Å pore size, Thermo Fisher Scientific). Peptides were eluted with a gradient of acetonitrile. The analytical column outlet was directly interfaced through a nano-flow electrospray ionization source, with a quadrupole Orbitrap mass spectrometer (Orbitrap Exploris 480, Thermo Fisher Scientific). MS data were acquired in data-dependent mode using a top ten method, where ions with a precursor charge state of 1+ and 2+ were excluded. High-resolution full scans (R = 60,000, m/z 380–1,800) were recorded in the Orbitrap followed by higher-energy collision dissociation (HCD) (stepped collision energy 30 and 32% normalized collision energy) of the ten most intense MS peaks. The fragment ion spectra were acquired at a resolution of 30,000 and a dynamic exclusion window of 20 s was applied.
For data analysis, Xcalibur raw files were converted into the MGF format using Proteome Discoverer v.2.3 (Thermo Fisher Scientific) and used directly as input files for MeroX78. Searches were performed against an ad hoc protein database containing the sequences of the proteins in the complex and a set of randomized decoy sequences generated by the software. The following parameters were set for the searches: maximum number of missed cleavages: 3; targeted residues K, S, Y and T; minimum peptide length 5 amino acids; variable modifications: carbamidomethylation of cysteine (mass shift 57.02146 Da), methionine oxidation (mass shift 15.99491 Da); DSBU modified fragments: 85.05276 Da and 111.03203 Da (precision: 5 ppm MS and 10 ppm MS/MS); false-discovery-rate cut-off: 5%. Finally, each fragmentation spectrum was manually inspected and validated.
To compare with the peptide array experiments, the number of cross-links detected for each amino acid residue was counted, and summed within an individual 20 amino acid peptide with a 1 amino acid shift, similar to the peptide array. The overlayered result was plotted using GraphPad Prism 9.
Peptide array
Peptides (20 amino acids) with 1-amino-acid shift covering the full sequences of RAD52, RPA1, RPA2 and RPA3 were synthesized on cellulose membranes in 3 mm spots by the Chemical Biology STP at the Francis Crick Institute. The membranes were washed with 50% ethanol and 10% acetic acid for 30 min and equilibrated with 1× TBST (50 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.1% Tween-20) supplemented with 0.25 mM TCEP. The membrane was blocked with 5% non-fat milk in TBST (0.1% Tween-20) supplemented with 0.25 mM TCEP for 1 h at room temperature. To allow protein-peptide interactions, the membranes were incubated with RAD52-OR or RPA (1 µg ml−1) in 1% non-fat milk in TBST (0.1% Tween-20) supplemented with 0.25 mM TCEP overnight at 4 °C. The membranes were washed in 1× TBST (0.1% Tween-20) supplemented with 0.25 mM TCEP on an orbital shaker for 5 min at room temperature three times. The membranes were then incubated in primary antibodies (anti-His 1:1,000, Takara, 631212) in 1% non-fat milk in TBST (0.1% Tween-20) supplemented with 0.25 mM TCEP for 2 h at room temperature. The membranes were washed three times as before and incubated in Alexa-Fluor-Plus-conjugated secondary antibodies (goat anti-mouse 1:2,000, Thermo Fisher Scientific, A32730; goat anti-rabbit, 1:2,000, Thermo Fisher Scientific, A32735) in 1% non-fat milk in TBST (0.1% Tween-20) supplemented with 0.25 mM TCEP for 1 h at room temperature. The membranes were washed three times, imaged on a Li-Cor Odyssey DLx system and quantified using Image Studio Lite (Li-Cor).
Nanoscale differential scanning fluorometry
A Prometheus NT-48 (Nanotemper) instrument was used to monitor changes in tryptophan fluorescence following thermal denaturation. Proteins were diluted to 10 µM in 25 mM HEPES pH 8.0, 200 mM KOAc, 0.5 mM EDTA, 10% glycerol and 0.25 mM TCEP. The samples were loaded into high-sensitivity glass capillaries and the tryptophan fluorescence was monitored at 330 and 350 nm after excitation at 285 nm. Measurements were made from 25 to 95 °C with a temperature gradient of 1 °C min−1. The ratio of fluorescence intensity (350/330 nm) was plotted against temperature, and the first derivative of this curve was used to calculate thermal melting (Tm) values.
Statistics and reproducibility
Statistical analyses were performed using GraphPad Prism 9. Normally distributed data were compared using two-tailed unpaired t-tests whereas non-normally distributed data were compared using two-tailed Mann–Whitney U-tests. Differences were considered to be statistically significant when P < 0.05. Reported n values refer to independent experiments for fluorescence anisotropy, biolayer interferometry analysis and SSA assays. Glycerol gradient sedimentation analysis and size-exclusion chromatography of U2OS nuclear extract recombinant RAD52-OR were repeated independently seven times with similar results. RAD52–ssDNA–RPA pull-down experiments were repeated independently five times with similar results. RAD52 purifications were repeated independently more than 50 times with similar results. RPA purifications were repeated for ten times with similar results. Purifications of RAD52 and RPA mutants were repeated for twice with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.