The enzyme cyclin-dependent kinase 1 (CDK1) and its partner cyclin B1 were thought to be sufficient to achieve error-free cell division. But now CDK5, an atypical cyclin-dependent kinase mostly known for its functions in non-dividing neurons, is shown to help to reduce errors in cell division.
Two changes make tomatoes tastier.Credit: Majdi Fathi/NurPhoto/Getty
Rotten tomatoes no more: growing sweeter tomatoes is possible by editing just two of the fruit’s genes. Deleting the genes increased the engineered fruits’ glucose and fructose levels by up to 30% over mass-produced tomatoes, according to a study1 published today in Nature.
Better yet, the gene-edited tomatoes weigh roughly the same as those sold now, and the plants produce as much fruit as do current varieties. These findings could not only help to improve tomatoes worldwide but are also an important step forward in understanding how fruits produce and store sugar, the authors write.
This study is “great and significant in its field and beyond”, says Christophe Rothan, a fruit biologist at the French National Institute of Agricultural Research in Paris, who was not involved in the study. It raises the “possibility of using the great genetic diversity existing in wild species, which has been partially lost in domesticated varieties, to improve modern varieties”, he says.
Special sauce
More than 186 million tonnes of tomatoes are produced globally each year, making the fruit one of the most valuable horticultural crops in the world. Like other crops, tomatoes have been domesticated by selecting for traits that reflect human preference — such as fruit size. Cultivated tomatoes today are up to 100 times larger than their wild ancestors, helping to boost how much fruit each plant produces.
Gene-edited tomatoes could provide new source of vitamin D
But this large size comes at a cost: typically, the bigger the fruit, the lower the proportion of the sugars that are responsible for the classic home-grown tomato taste. Supermarket tomatoes, by contrast, “taste like water”, says study co-author Jinzhe Zhang, a plant geneticist at the Chinese Academy of Agricultural Sciences in Beijing. “They’re flavourless.”
To address this problem, Zhang and his colleagues compared the genomes of cultivated tomato species (Solanum lycopersicum) with their much-sweeter wild counterparts. They found the sweet spot in two genes, each encoding a protein that degrades enzymes responsible for sugar production. Using CRISPR–Cas9 gene-editing technology, the researchers deactivated the two genes and found that the plants bore fruit that was much sweeter than that of a widely cultivated variety.
The new tomato would be welcome not only because it would make consumers happy, but also because it could cut the amount of time, energy and money that goes into preparing other products such as tomato paste, which involves removing water from the fruit, says Ann Powell, a retired plant biochemist who previously worked at the University of California, Davis.
The findings might bear fruit for other produce as well too: these genes are found across a range of plant species, and the mechanisms that underlie sugar production in fruits have long stumped scientists, Powell says.
In September, a man from Montana was sentenced to six months in prison after he trafficked a clone of one of the world’s largest sheep species. Court documents allege that Arthur Schubarth trafficked body parts of a near threatened Marco Polo argali sheep into the US from Kyrgyzstan and in 2015 contracted with a lab to create a cloned sheep he later named Montana Mountain King (MMK). Later, the documents allege, Schubarth used MMK’s semen to impregnate ewes and then sold offspring—each carrying some Marco Polo argali genetics—to people involved in big game hunting.
It’s a weird case. It’s likely only the second time that an American has been prosecuted for a wildlife crime that involved animal cloning. (In 2011 a man was fined $1.5 million and ordered to surrender smuggled deer as well as nearly $1 million of deer semen—which investigators believed he intended to use to clone whitetail deer—in a case that involved the unlawful purchase and transportation of deer.)
There’s another strange element to Schubarth’s story: Potentially dozens of MMK’s descendants may now be at large in the US. These sheep that contain genetics from MMK are defined as contraband in the handful of plea agreements that were signed by men who were alleged to have bought sheep from Schubarth or transported ewes to his ranch in Montana to be impregnated. What isn’t clear is how many sheep are at large, and what exactly has happened to them.
However, legal documents offer some clues. One legal filing in the case against Schubarth alleges that in November 2018 one person transported 26 ewes to Schubarth’s ranch in Montana to be inseminated with MMK semen, and a year later the same person later transported another 48 ewes. In July 2020, the same document alleges, two other people transported another 43 sheep to Schubarth’s ranch. That’s at least several dozen sheep that may have carried MMK’s offspring—and each of those may have had several lambs.
The same document also alleges that one of MMK’s offspring was transported from Minnesota to Schubarth’s ranch in Montana in May 2019. Then in July 2020 Schubarth agreed to sell 11 of MMK’s grandchildren for a total of $13,200 and one of MMK’s children, a sheep called Montana Black Magic, for $10,000. It’s also alleged that Schubarth sold another Marco Polo hybrid sheep to a man who lives in South Dakota.
At least one sheep is accounted for: MMK himself. The sheep had initially been taken to a Zoological Association of America accredited facility in Oregon, says Christina Meister of the US Fish and Wildlife Service (USFWS) Office of Public Affairs. On October 2, MMK was flown across the country to Rosamond Gifford Zoo in Syracuse, New York, where he will be housed for the long term. MMK is expected to be on exhibition at the zoo in mid-November, Meister says. (The USFWS declined to answer other questions posed by WIRED.)
RNA can adopt many configurations in the cell (artist’s illustration).Credit: Christoph Burgsted/Science Photo Library/Getty
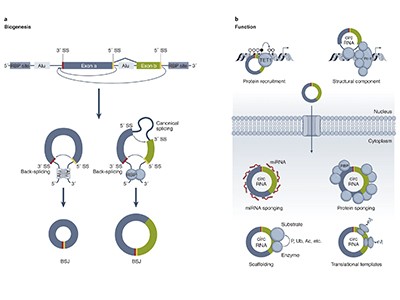
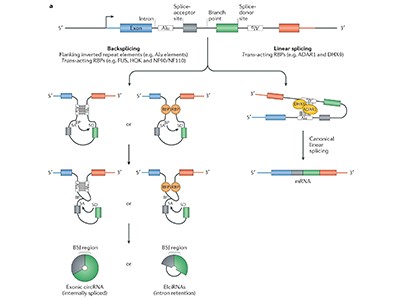
Over the past few decades, RNA’s place in biology has transformed from being a mere intermediate between DNA and protein to a fascinating molecule with diverse activities that go well beyond simple transcription of genetic information. Many of these RNAs fold up like molecular origami, but one of their most puzzling configurations is circular: molecules in which an unusual version of the standard RNA-splicing process folds the strand back on itself, creating a loop.
Once thought to be artefacts of splicing gone wrong, circular RNAs (circRNAs) are now known to be widespread across the tree of life. They’ve been implicated in conditions including cancer, cardiovascular disease and Alzheimer’s disease, and offer exciting possibilities as both therapeutic agents and biomarkers.
Even so, scientists are still working out what these molecules do. Some of the first circRNAs to be studied mop up small, non-coding RNAs called microRNAs to prevent them from binding to messenger RNAs and dampening protein production. But others might interact with various proteins or the enzyme RNA polymerase to regulate transcription and protein expression, or even be translated themselves (see ‘Potential functions of circular RNA’).
Source: V. M. Conn et al. Nature Rev. Cancer24, 597–613 (2024).
“There are really very interesting things to be found out about circular RNAs,” says Nikolaus Rajewsky, a systems biologist at the Max Delbrück Center for Molecular Medicine in Berlin. “Largely, it’s terra incognita, and that’s very exciting.”
Not to mention challenging. That’s in part because circRNAs are rare — they make up about 0.1% of non-ribosomal RNA sequences, by one measurement1 — and also because they’re essentially identical to linear RNAs transcribed from the same part of the genome. Their only distinguishing feature is where they’re joined together to form a circle, that is, where the end of one RNA segment links up with a segment from earlier in the DNA code. This is called the back-splice junction. It’s difficult to analyse or generate pure circular forms without the linear ones interfering, and specialists warn that the scientific literature is littered with spurious findings that link a particular circRNA with a specific microRNA without iron-clad evidence. “You really need to do a thousand controls,” says Rajewsky.
Researchers who are new to the field can follow published best practices2,3. But the ideal approach is to consult with circRNA veterans before starting a project, suggests Grace Chen, an RNA biologist at Yale University School of Medicine in New Haven, Connecticut. “Talk to us early, and talk to us often,” she says.
That said, researchers are adapting a variety of methods from standard RNA biology to identify and investigate these exciting molecules, and the circRNA-specific toolbox is growing. “I think people should do the hard work to get into this field if this is something that they’re interested in,” says Chen. “There’s a lot to be discovered here.”
Loopy molecules
Step one is to identify circRNAs of interest. There are multiple databases for this, but many are uncurated, incomplete and riddled with unvalidated listings, cautions Jo Vandesompele, a cancer-genomics researcher at Ghent University in Belgium who reviewed4 circRNA databases with his colleagues in 2020. The naming system is a jungle, he says, with many names for the same molecule, and the database landscape a full-scale nightmare. If he had to pick one database, he says, it would be circAtlas from the Beijing Institutes of Life Science in China. That’s because circAtlas requires any listed circRNAs to be identified by two tools, each with at least two counts of the back-splice junction.
Why rings of RNA could be the next blockbuster drug
Sequencing is the most common approach to find new circRNAs, but these loopy molecules are often missing from standard RNA libraries. Scientists usually build these libraries by targeting the poly(A) tail attached to mRNAs — a feature that circRNAs lack. “You need to be specifically looking for them,” says Jeremy Wilusz, an RNA biologist at Baylor College of Medicine in Houston, Texas.
More specifically, circle hunters must seek out that back-splice junction. It is not a single signature sequence, but any place where the 3′ end of one coding sequence, or exon, connects to the 5′ end of an exon that is normally upstream in the linear sequence — and non-coding sequences called introns can also get in on the act.
The most common option is to create total RNA libraries, says Vandesompele: it’s easier and less expensive than making circRNA-specific libraries, and means scientists can look for multiple RNA forms, not just circles. First, however, they have to get rid of ribosomal RNA, a structural component of ribosomes that represents the vast majority of RNAs in cells.
Given the low abundance of most circRNAs, getting enough material to sequence is part of the challenge, says Prisca Obi, an immunobiologist and newly minted PhD graduate in Chen’s laboratory at Yale University. The cost of commercial kits to eliminate ribosomal RNA would quickly add up before scientists obtained much circRNA. So, Chen and her colleagues developed an inexpensive, do-it-yourself protocol5. They add DNA probes containing sequences that match ribosomal RNA, then use the enzyme RNase H to destroy the resulting RNA–DNA hybrid molecules. After a few further clean-up steps, “you should have RNA that’s mostly depleted of ribosomal RNA”, says Obi.
The alternative is to strip away most of the non-circular RNAs, too. This requires the enzyme RNase R, which attacks linear RNAs at their ends. But it’s important to titrate the reaction conditions carefully, Vandesompele warns: too much enzyme will destroy the circles, and too little will spare some of the linear pieces. Also, certain RNA structures, such as G-quadruplexes, histone mRNAs and small nuclear RNAs, can foil the enzyme. Swapping the potassium in the reaction buffer for lithium destabilizes these structures and helps to improve the degradation of linear RNAs, Wilusz and a colleague have reported6.
Telltale junctions
Once researchers have sequenced the RNAs — short-read methods such as those developed by US biotechnology firm Illumina are generally fine, says Vandesompele — the next step is to search those sequences for the telltale back-splice junctions . But the algorithms available for doing so vary widely in sensitivity.
In a 2023 comparison of 16 tools conducted by Vandesompele and his colleagues, individual algorithms identified anywhere from 1,372 to 58,032 circRNAs from the same cell line7. Vandesompele recommends researchers select at least two tools that have low false-positive rates; his lab has landed on CirComPara2 and circtools, but others might have different needs.
Best practice standards for circular RNA research
None of these algorithms is perfect, Rajewsky notes: “In our experience, about 80% of these things are real.” Unexpected splice forms or amplification issues can create spurious results.
Another catch is that most search algorithms rely on matching RNA sequences to a reference genome, says Julia Salzman, a computational biologist at Stanford University in California. If the circRNA sequence is missing from the reference genome — because, say, you’re searching for a viral circRNA but the reference sequence is human — it will never be found, creating a false-negative result. And because near-homologous sequences are found throughout the genome, attempts to match the sequences can also create false positives.
Salzman and her colleagues developed an alternative approach called SPLASH2. The software compares two sets of sequences without relying on a reference genome, on the basis of counts of short genetic segments called k-mers. Salzman recommends comparing a sample containing both circles and linear RNAs with one treated with RNase R to reduce the linear component — differences between the two samples will point to circRNAs. In one specificity test, 92% of potential circRNAs identified by SPLASH2 were known or likely circles8.
Circular validation
To quantify recognized circRNAs, scientists can use microarray chips studded with nucleic-acid probes for known circRNA back-splice junctions. Scientists can wash their samples over the chip to detect when a sequence in the sample matches its partner on the array.
Arraystar, a biotechnology company focused on non-coding RNAs in Rockville, Maryland, has designed circRNA microarrays targeting the known circles for human, mouse and rat samples, and researchers have used them, among other things, to identify a circRNA in mouse blood stem cells that helped the cells to avoid exhaustion9. A key caveat: “If it’s not there on the array, it could be super-interesting, and you’ll never find it,” says Wilusz.
Yanggu Shi, a senior scientist at Arraystar, says that about 70% of microarray predictions hold up. So, the next step for any detection approach is to validate that the circRNAs are present and genuinely circular — typically by using quantitative PCR. Researchers treat part of a sample with RNase R to degrade linear RNAs and leave part untreated, and amplify both samples; any real circles should withstand the enzyme treatment, whereas linear counterparts would be diminished.
That said, the back-splice junction represents just one part of the RNA: there could be several circles, each with different complements of exons or even introns sharing the same junction. That’s why Wilusz’s validation strategy includes non-PCR approaches, such as Northern blotting, in which researchers separate RNA molecules by size and then use sequence-specific probes to detect molecules of interest. That way, they can not only detect specific RNAs, but can also determine how many molecules of different sizes contain those sequences. Long-read sequencing, although expensive, is another option. “Only then can you have an unambiguous identification,” says Vandesompele.
The biogenesis, biology and characterization of circular RNAs
The abundance of a circRNA can provide an important clue to its function, says Vanessa Conn, a molecular biologist at Flinders University College of Medicine and Public Health in Bedford Park, Australia. For example, if scientists predict that a circRNA is mopping up microRNAs, then the circRNA should be sufficiently abundant to catch most of the microRNAs in a cell. But even a rare circRNA might be able to influence a gene at the DNA level, because there are only two copies of it in the genome. For example, Conn, together with her husband Simon Conn, a molecular cancer biologist who leads a lab at Flinders University, discovered one low-level circRNA that interacts with DNA to drive genetic translocations associated with leukaemia10.
Quantifying circRNAs can be tricky, the Flinders researchers say. That’s because small circRNAs in a sample will be amplified more quickly than are large ones, making the small molecules seem more abundant than they are. Conn and his group developed a method they call SplintQuant to get around that problem11. Once scientists have identified a circle of interest, they can design DNA probes to match either side of its back-splice junction. Then they use an enzyme to link up any pairs of probes that have found a circle together, and use quantitative PCR to count the ligated molecules. But what about comparing circRNA sequences and quantities between labs and protocols? The Conns propose a solution here, too, in the form of synthetic circRNAs called SynCRS (pronounced sinkers) that researchers can spike into their samples before library production12. Using known quantities of SynCRS allows scientists to normalize results across labs.
Muddied waters
From there, scientists can finally move on to the most interesting challenge: function. Key approaches include knocking down or overexpressing the circles, and again, similarities to other RNA species muddy the waters.
A technique called RNA interference is one common approach, because it can be targeted to the back-splice junction, says Guillermo Aquino-Jarquin, a researcher in medical sciences at the Federico Gómez Children’s Hospital of Mexico in Mexico City. For example, by targeting the cancer-linked circRNA circAGO2 in mice, using small hairpin RNAs, researchers demonstrated the gene’s role in promoting tumour generation and aggressiveness13. Alternatively, if researchers suspect that specific circle sequences have binding partners among the nucleic acids or proteins in a cell, they can design interfering RNAs to bind to and suppress those sites specifically.
The alternative is to use the CRISPR–Cas system to target and obliterate circles of interest. With Cas9, a DNA-targeting enzyme, researchers can damage a circRNA gene itself, destroying its ability to make the molecules. Rajewsky and colleagues used this strategy to make mice deficient in the circRNA Cdr1as, which interacts with the microRNA miR-7. Without Cdr1as, cells contained less miR-7, suggesting that the circRNA controls the microRNA’s stability or perhaps its transport14.
That said, linear RNA production can also be affected by alterations to the genome. Alternatively, researchers can use the enzyme Cas13 to target the back-splice junction or other binding sites at the RNA level15. “You knock down the circRNA, but you don’t touch the genome,” says Aquino-Jarquin. Knockdowns of this type are about 80–90% effective, he estimates, compared with 50–60% effectiveness for RNA interference. Cas enzymes can also create off-target effects, however.
NatureTech hub
Thus, as with circularity itself, validation and controls remain key, says Wilusz. For example, researchers who think that a circRNA might be sponging up microRNAs might eliminate the circle itself, as well as mutating likely microRNA binding sites and looking for similar results. And, if eliminating a circle produces a phenotype, adding that circle back in should reverse it. That’s where overexpression comes in. This is doable, but impossible to achieve perfectly, says Rajewsky. “Nobody can produce, as far as I know, 100% clean, circular RNAs,” he says. “You will also introduce some other RNAs.”
Researchers can generate circRNAs synthetically or make them in vivo using plasmids that are designed to use the cell’s own splicing machinery. The latter approach “would, ideally, most faithfully recapitulate the cell making it”, says Obi. One classic approach, the permuted intron–exon (PIE) strategy, involves rearranging exons and introns into configurations that promote automatic release of a circularized product16. Plasmids that generate circRNAs are available from the US non-profit plasmid repository Addgene.
For the best control over circRNA purity, synthetic benchtop methods are preferable, Obi says. She and Chen like to generate linear RNA precursors, then link them together with an enzyme called ligase. To minimize undesirable side products, researchers can add RNase R to degrade unwanted linear RNAs, and use separation techniques such as gel electrophoresis or liquid chromatography to purify the desired species.
But as always, Rajewsky warns, there are caveats. With synthetic circles, “whatever you’re putting in is artificial, and might not relate at all to the biology that’s happening in the cells.”
So when it comes to circRNAs, the key is to test hypotheses with multiple methods, he says. For example, Rajewsky and his colleagues recently reported that the circRNA Cdr1as interacts with miR-7 to regulate the release of the neurotransmitter glutamate from neurons17. They used a variety of techniques: mammal primary neuron culture, chemical and electrical stimulation, single-cell RNA imaging and microRNA knockouts — to name a few.
Is that overkill? Not when it comes to circRNAs, says Rajewsky. “It is necessary to really say something substantial about these crazy molecules.”
DNA samples from one of the world’s largest and oldest plants — a quaking aspen tree (Populus tremuloides) in Utah called Pando — have helped researchers to determine its age and revealed clues about its evolutionary history.
By sequencing hundreds of samples from the tree, researchers confirmed that Pando is between 16,000 and 80,000 years old, verifying previous suggestions that it is among the oldest organisms on Earth. They were also able to track patterns of genetic variation spread throughout the tree that offer clues about how it has adapted and evolved over the course of its lifetime. The findings were posted on the bioRxiv preprint server on 24 October1. The work has not yet been peer reviewed.
“It’s just pretty cool to study such an iconic organism,” says co-author Rozenn Pineau, a plant evolutionary geneticist at the University of Chicago in Illinois. “I think it’s important to draw people’s attention on natural wonders of the world.”
One very big tree
Pando — whose name means ‘I spread’ in Latin — consists of some 47,000 stems that cover an area of 42.6 hectares in Utah’s Fishlake National Forest. Because of the way the plant reproduces, this collection of aspens is technically all one tree, supported by a single, vast root system. Pando is triploid, meaning that its cells contain three copies of each chromosome, rather than two. As a result, Pando cannot reproduce sexually and mix its DNA with that of other trees, and instead creates clones of itself.
The trees’ lessons: climate records are written in tree rings
Although this process generates offspring that are genetically identical, they can still accumulate genetic mutations as their cells divide. Biologists are interested in these variations because they provide information on how the plant has changed since the first seedling sprouted. Some studies have explored the spread of new mutations in plants and fungi that reproduce clonally, but few have investigated centuries-old plants like Pando.
“It’s kind of shocking to me that there hasn’t been a lot of genetic interest in Pando already, given how cool it is,” says study co-author William Ratcliff, an evolutionary biologist at the Georgia Institute of Technology in Atlanta.
The researchers collected samples of roots, bark, leaves and branches from across the Pando clone, as well as from other, unrelated quaking aspen trees for comparison. They extracted DNA from the samples, then sequenced and analysed a subsection of the genome.
After removing variants that were found in both Pando and neighbouring trees, as well as mutations found in just one sample, the researchers were able to review nearly 4,000 genetic variants that had arisen as Pando cloned itself repeatedly over millennia.
Analysing the patterns of these mutations revealed surprising results. “You would expect that the trees that are spatially close are also closer genetically,” says Pineau. “But this is not exactly what we find. We found a spatial signal, but that is much weaker than what we expected.” Physically close trees did share more similar mutations than those that were far apart — but only slightly more. However, over a smaller scale of 1–15 metres the trend was stronger, with stems that were closer together having significantly more shared mutations. Pando covers an area of more than 40 hectares, “but it almost looks like it’s a well-mixed pot of genetic information”, says Ratcliff.
Protective mechanism
By inputting Pando’s genetic data into a theoretical model that plots an organism’s evolutionary lineage, the researchers also estimated Pando’s age. They put this at between 16,000 and 80,000 years. “It makes the Roman Empire seem like just a young, recent thing,” says Ratcliff.
The team also considered reasons for the tree’s remarkable endurance. Pineau says that Pando being triploid might lead to “bigger cells, bigger organisms, better fitness”, and that existing clones might be more durable than new mixed offspring.
Philippe Reymond, who researches interactions between plants and herbivores at the University of Lausanne in Switzerland, says that the findings hint that “plants and trees have a mechanism to protect the genome” from the accumulation of harmful genetic mutations, a suggestion that is “quite interesting for many scientists”. He adds that future studies could search for this exact mechanism at the cellular level.
Ratcliff is also keen for more studies to be done on Pando’s genetic history. “I would love to make a call for people to work on these kinds of organisms,” he says.
It had long been suspected that the occurrence of breast cancer had a familial component, but early studies were confounded by the complexity of the disease. However, in 1990, researchers identified a key genetic determinant of inherited breast cancer risk1, which fired the starting gun for what turned out to be a four-year race to identify the underlying gene. This was achieved when Miki et al., in a 1994 paper in Science2, described a previously unknown gene in which affected individuals in high-risk families carried deleterious mutations. The work provided strong evidence that this was indeed the BRCA1 gene.
Competing Interests
A.A. is a co-founder of Tango Therapeutics, Azkarra Therapeutics, Ovibio, Kytarro and TillerTx; a member of the board of Cytomx, Ovibio Corporation, Cambridge Science Corporation; a member of the scientific advisory board of Genentech, GLAdiator, Circle, Bluestar/Clearnote Health, Earli, Ambagon, Phoenix Molecular Designs, Yingli/280Bio, Trial Library, ORIC and HAP10; a consultant for ProLynx, Next RNA and Novartis; receives research support from SPARC; and holds patents on the use of PARP inhibitors held jointly with AstraZeneca from which he has benefited financially (and may do so in the future).
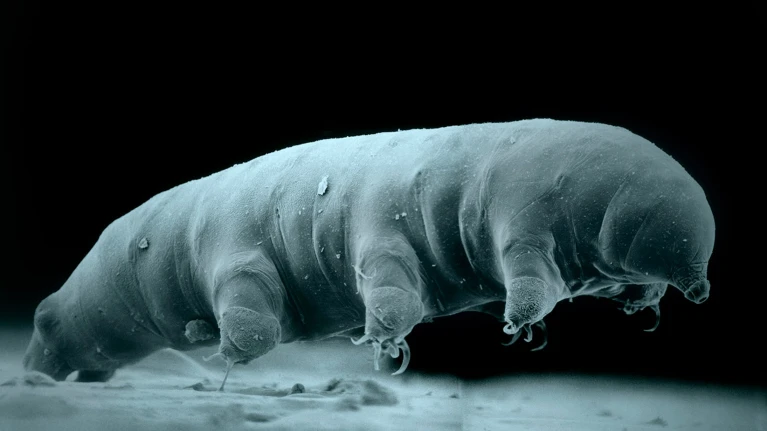
The new species is similar in appearance to this Hypsibius sp. tardigrade, photographed using an electron microscope.Credit: Robert Schuster/Science Photo Library
A newly described species of tardigrade is giving scientists insights into what makes these tiny eight-legged creatures so resistant to radiation.
Tardigrades, also known as water bears, have long fascinated scientists with their ability to withstand extreme conditions, including radiation at levels nearly 1,000 times higher than the lethal dose for humans. There are around 1,500 known tardigrade species, but only a handful are well-studied.
See a tardigrade ride a worm in the world’s weirdest rodeo — September’s best science images
Now, scientists have sequenced the genome of a species new to science, and revealed some of the molecular mechanisms that give tardigrades their extraordinary resilience. Their study, published in Science on 24 October1, identifies thousands of tardigrade genes that become more active when exposed to radiation. These processes point to a sophisticated defence system that involves protecting DNA from the damage that radiation causes and repairing any breaks that do occur.
The authors hope that their insights could be harnessed to help protect astronauts from radiation during space missions, clean up nuclear pollution or improve cancer treatment.
“This discovery may help improve the stress tolerance of human cells, benefiting patients undergoing radiation therapy,” says study co-author Lingqiang Zhang, a molecular and cellular biologist at the Beijing Institute of Lifeomics.
Protective genes
Around six years ago, Zhang and his colleagues ventured into Funiu Mountain in China’s Henan province to collect moss samples. Back in the laboratory and under the microscope, they identified a previously undocumented species of tardigrade, which they named Hypsibius henanensis. Genome sequencing revealed that the species had 14,701 genes, 30% of which are unique to tardigrades.
When the researchers exposed H. henanensis to radiation doses of 200 and 2,000 grays — far beyond what would be survivable for humans — they found that 2,801 genes involved in DNA repair, cell division and immune responses became active.
“It’s like when in wartime, when factories are refitted to just make munitions. It’s almost that level of retooling how gene expression is working,” says Bob Goldstein, a cell biologist at the University of North Carolina at Chapel Hill, who has been studying tardigrades for 25 years. “We’re fascinated by how an organism would change their gene expression to a point where they’re making that much transcript for specific genes.”
What Chernobyl’s stray dogs could teach us about radiation
One of the genes, called TRID1, encodes a protein that helps to repair double-strand breaks in DNA by recruiting specialized proteins at sites of damage. “This is a new [gene] that, to my knowledge, no one was studying,” says Goldstein.
The researchers also estimate that 0.5–3.1% of the tardigrade’s genes were acquired from other organisms through a process known as horizontal gene transfer. A gene called DODA1, which seems to have been acquired from bacteria, enables tardigrades to produce four types of antioxidant pigments called betalains. These pigments can mop up some of the harmful reactive chemicals that radiation causes to form inside cells, which account for 60–70% of radiation’s damaging effects.
The authors treated human cells with one of the tardigrade’s betalains and found that they were much better at surviving radiation than cells that were not treated.
No expiration date
Studying the molecular mechanisms that allow tardigrades to tolerate other harsh conditions, such as extreme temperatures, air deprivation, dehydration and starvation, could have wide applications. It could improve the shelf life of fragile substances such as vaccines, for example. “All your medicines have expiration dates — tardigrades don’t,” says Goldstein.
Comparing these mechanisms between different tardigrades is an important part of this research, adds Nadja Møbjerg, an animal physiologist at the University of Copenhagen. “We are still lacking knowledge of different tardigrade species out there,” she says.
These animals have “a font of protectants that will probably keep spilling out more that will be useful and interesting to understand”, says Goldstein. “We want to understand how those work and what potential they have.”
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature562, 203–209 (2018). This paper provides a broad overview of the population-based UK Biobank sample, which has had a transformative influence on epidemiology and the genetic study of complex traits.
Davies, N. M., Dickson, M., Davey Smith, G., van den Berg, G. J. & Windmeijer, F. The causal effects of education on health outcomes in the UK Biobank. Nat. Hum. Behav.2, 117–125 (2018).
Yengo, L. et al. Imprint of assortative mating on the human genome. Nat. Hum. Behav.2, 948–954 (2018). This paper introduced an approach to test the extent of assortative mating across traits using molecular genetic data.
Sanjak, J. S., Sidorenko, J., Robinson, M. R., Thornton, K. R. & Visscher, P. M. Evidence of directional and stabilizing selection in contemporary humans. Proc. Natl Acad. Sci. USA115, 151–156 (2018).
Abdellaoui, A., Yengo, L., Verweij, K. J. H. & Visscher, P. M. 15 years of GWAS discovery: realizing the promise. Am. J. Hum. Genet.110, 179–194 (2023).
Lawlor, D. A. & Mishra, G. D. (eds) Family Matters: Designing, Analysing, and Understanding Family-Based Studies in Life Course Epidemiology (Oxford Univ. Press, 2009).
Dicks, A., Levels, M., van der Velden, R. & Mills, M. C. How young mothers rely on kin networks and formal childcare to avoid becoming NEET in the Netherlands. Front. Sociol.6, 787532 (2021).
Bratti, M., Fiore, S. & Mendola, M. The impact of family size and sibling structure on the great Mexico–USA migration. J. Popul. Econ.33, 483–529 (2020).
Chetty, R., Hendren, N., Kline, P. & Saez, E. Where is the land of opportunity? The geography of intergenerational mobility in the United States. Q. J. Econ.129, 1553–1623 (2014).
Taubes, G. Epidemiology faces its limits: the search for subtle links between diet, lifestyle, or environmental factors and disease is an unending source of fear—but often yields little certainty. Science269, 164–169 (1995).
D’Onofrio, B. M., Lahey, B. B., Turkheimer, E. & Lichtenstein, P. Critical need for family-based, quasi-experimental designs in integrating genetic and social science research. Am. J. Public Health103, S46–S55 (2013).
Cnattingius, S. The epidemiology of smoking during pregnancy: Smoking prevalence, maternal characteristics, and pregnancy outcomes. Nicotine Tob. Res.6, 125–140 (2004).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet.11, 459–463 (2010).
Young, A. I. et al. Mendelian imputation of parental genotypes improves estimates of direct genetic effects. Nat. Genet.54, 897–905 (2022). This paper demonstrates that missing genotypes of relatives can be imputed in a way that provides unbiased estimates of direct and indirect genetic effects.
Howe, L. J. et al. Within-sibship genome-wide association analyses decrease bias in estimates of direct genetic effects. Nat. Genet.54, 581–592 (2022). This paper used a large sample of siblings to estimate direct genetic effects and to demonstrate that genetic associations are inflated in samples of unrelated individuals for many traits.
Mathieson, I. & McVean, G. Differential confounding of rare and common variants in spatially structured populations. Nat. Genet.44, 243–246 (2012). This paper demonstrated that principal components can control for population stratification of common variants but that this approach is less successful for rare variants.
Persyn, E., Redon, R., Bellanger, L. & Dina, C. The impact of a fine-scale population stratification on rare variant association test results. PLoS ONE13, e0207677 (2018).
Kong, A. et al. The nature of nurture: effects of parental genotypes. Science359, 424–428 (2018). This paper was the first to demonstrate indirect genetic effects using molecular genetic data in very large samples of trios.
Demange, P. A. et al. Estimating effects of parents’ cognitive and non-cognitive skills on offspring education using polygenic scores. Nat. Commun.13, 4801 (2022).
Wang, B. et al. Robust genetic nurture effects on education: A systematic review and meta-analysis based on 38,654 families across 8 cohorts. Am. J. Hum. Genet.108, 1780–1791 (2021).
Young, A. I., Benonisdottir, S., Przeworski, M. & Kong, A. Deconstructing the sources of genotype-phenotype associations in humans. Science365, 1396–1400 (2019).
Border, R. et al. Assortative mating biases marker-based heritability estimators. Nat. Commun.13, 660 (2022). This paper reports the extent of cross-trait assortative mating and its implications for misinterpretations of genetic correlations.
Van Der Laan, J., De Jonge, E., Das, M., Te Riele, S. & Emery, T. A whole population network and its application for the social sciences. Eur. Sociol. Rev.39, 145–160 (2023).
Liu, A. et al. Evidence from Finland and Sweden on the relationship between early-life diseases and lifetime childlessness in men and women. Nat. Hum. Behav.8, 276–287 (2023).
Allesøe, R. L. et al. Deep learning for cross-diagnostic prediction of mental disorder diagnosis and prognosis using Danish nationwide register and genetic data. JAMA Psychiatry80, 146 (2023).
Boyd, A. et al. Cohort profile: The ‘Children of the 90s’—the index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol.42, 111–127 (2013).
Power, C., Kuh, D. & Morton, S. From developmental origins of adult disease to life course research on adult disease and aging: insights from birth cohort studies. Annu. Rev. Public Health34, 7–28 (2013).
Larmuseau, M. H. D. et al. Low historical rates of cuckoldry in a Western European human population traced by Y-chromosome and genealogical data. Proc. R. Soc. B Biol. Sci.280, 20132400 (2013).
Tomkins, S. in Family Matters: Designing, Analysing and Understanding Family Based Studies in Life Course Epidemiology (eds Lawlor, D. A. & Mishra, G. D.) Ch. 8, 129–150 (Oxford Univ. Press, 2009).
Berthoud, R., Fumagalli, L., Lynn, P. & Platt, L. Design of the Understanding Society Ethnic Minority Boost Sample.Working Paper No. 2009-02 (Institute for Social and Economic Research, University of Essex, 2009).
Schreuder, P. & Alsaker, E. The Norwegian Mother and Child Cohort Study (MoBa) – MoBa recruitment and logistics. Nor. Epidemiol.24, 23–27 (2014).
Fry, A. et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am. J. Epidemiol.186, 1026–1034 (2017).
Lawlor, D. A. & Leon, D. A. in Family Matters: Designing, Analysing and Understanding Family Based Studies in Life Course Epidemiology (eds Lawlor, D. A. & Mishra, G. D.) Ch. 13, 263–278 (Oxford Univ. Press, 2009).
Davies, N. M., Holmes, M. V. & Smith, G. D. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Brit. Med. J.362, k601 (2018).
Brumpton, B. et al. Avoiding dynastic, assortative mating, and population stratification biases in Mendelian randomization through within-family analyses. Nat. Commun.11, 3519 (2020).
Howe, L. J. et al. Educational attainment, health outcomes and mortality: a within-sibship Mendelian randomization study. Int. J. Epidemiol.52, 1579–1591 (2023).
Peterson, R. E. et al. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell179, 589–603 (2019).
Kendler, K. S., Gardner, C. O. & Lichtenstein, P. A developmental twin study of symptoms of anxiety and depression: evidence for genetic innovation and attenuation. Psychol. Med.38, 1567–1575 (2008).
Visscher, P. M. et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet.2, e41 (2006).
Balbona, J. V., Kim, Y. & Keller, M. C. Estimation of parental effects using polygenic scores. Behav. Genet.51, 264–278 (2021). This paper described how samples of related individuals with molecular genetic data can be used to estimate parental effects while controlling for assortative mating.
Sasani, T. A. et al. Large, three-generation human families reveal post-zygotic mosaicism and variability in germline mutation accumulation. eLife8, e46922 (2019).
Genomics England Research Consortium. Heritability of de novo germline mutation reveals a contribution from paternal but not maternal genetic factors. Preprint at bioRxivhttps://doi.org/10.1101/2022.12.17.520885 (2022).
Stankovic, S. et al. Genetic links between ovarian ageing, cancer risk and de novo mutation rates. Nature633, 608–614 (2014).
The late, great evolutionary biologist William Hamilton apparently used to correspond on second-hand postcards, writing over the original script in different-coloured ink, sometimes at right angles. In The Genetic Book of the Dead: A Darwinian reverie, his colleague Richard Dawkins describes this as a kind of palimpsest – “a manuscript in which later writing has been superimposed on earlier (effaced) writing”. This, he says, is a…