[ad_1]
Antibiotics can reduce diversity in the gut microbiome, raising the risk of infections that cause diarrhoea – and the effects may last years
[ad_2]
Source link


[ad_1]
Antibiotics can reduce diversity in the gut microbiome, raising the risk of infections that cause diarrhoea – and the effects may last years
[ad_2]
Source link

[ad_1]

It is one of the scourges of life in the modern world: chronic inflammation. This unhelpful response by the body’s immune system is linked to accelerated ageing and conditions such as stroke and heart disease.
What if we could dampen it down by consuming certain foods, such as spinach, walnuts and salmon? That is the promise of anti-inflammatory diets, often advocated in vague terms by the media and nutrition industry. That might prompt eye-rolling from the scientifically minded. But recent research reveals that this approach isn’t as faddish as it sounds and paints a nuanced picture of the links between food, inflammation and our long-term health.
Inflammation is a crucial part of our response to injury and disease. But when the body continues to deploy it even when there is no trauma, this results in chronic inflammation. Exactly why this occurs is unclear, but genetics, environment and lifestyle play roles. It can be detected by measuring certain chemical markers in the blood, and has been increasingly linked with poor health.
“Chronic inflammation is a driver of many common diseases, including cardiovascular disease, cancer, arthritis and dementia,” says John Mathers at Newcastle University in the UK. It has also been implicated in some mental health conditions.
But how…
[ad_2]
Source link

[ad_1]

Endometriosis tissue viewed under a microscope
BIOPHOTO ASSOCIATES/SCIENCE PHOTO LIBRARY
Low levels of a particular compound in faeces could be a sign of endometriosis – and supplementation of that compound might even help control the condition.
Affecting nearly 200 million people worldwide, endometriosis occurs when the tissue lining the uterus grows in other parts of the reproductive tract. There is no known cure, but lesions can be periodically removed through surgical procedures once the condition has been diagnosed. However, due in large part to a lack of awareness and understanding, it currently takes an average of more than six years for endometriosis to be diagnosed.
Previous research has suggested that the gut microbiome might play a role in the condition. To investigate further, Ramakrishna Kommagani at Baylor College of Medicine in Houston, Texas, and his colleagues collected stool samples from 18 women with endometriosis and 31 women without the condition. They investigated the bacteria in the faeces as well as the metabolome – the set of chemicals produced by the gut bacteria.
They found that the women with endometriosis had lower levels of the metabolite 4-hydroxyindole in their faeces, possibly due to alterations in the gut microbiome.
Based on that discovery, commercial stool analyses could allow rapid screening for this widely “underdiagnosed, understudied and underappreciated” condition, leading to early and effective management, says Kommagani.
“Stool is so easy to collect, and it’s not invasive like current diagnostic techniques such as laparoscopy [a kind of keyhole surgery],” he says.
To explore whether 4-hydroxyindole might even have a protective effect, the team fed supplementary 4-hydroxyindole to a group of mice that had tissue implanted in their abdomens to induce endometriosis. After 14 days of treatment, those mice did not have fewer lesions compared with control animals, but their lesions were remarkably less severe, and they showed signs of having significantly less pain.
Further experiments indicated that when mice with established endometriosis received 4-hydoxyindole, their lesions vastly improved. The results were similar in mice that had been grafted with human endometriosis lesions, suggesting the treatment could be effective in humans as well.
“We believe this is a very good therapeutic option, because it’s naturally occurring in the body – not a drug or something synthesised,” says Kommagani.
However, larger studies in humans will be needed to confirm whether 4-hydroxyindole can be used to diagnose endometriosis and whether the compound is effective as a treatment.
Topics:
[ad_2]
Source link

[ad_1]

In 2015, Nikki Schultek was in her prime: a young mother of two little boys, she had just run a half marathon. Then, a mysterious illness hit. Her asthma, previously well-managed, became increasingly severe. Over the following months, she experienced chronic pain, digestive problems and a cardiac arrhythmia. Then came the “last insult”: signs of neurodegeneration, including brain fog and lapses of memory. “It was the lowest point,” she recalls. “I began making plans for my kids, writing down notes of things that I would want to tell them if I continued to get worse.”
Schultek received various diagnoses for individual problems, but none fully matched her constellation of symptoms. Eventually, one doctor suggested that an undetected infection could lie behind her chronic pain and breathing difficulties. She tested positive for Borrelia burgdorferi and Chlamydia pneumoniae infections and was prescribed a cocktail of antibiotics. On taking them, she found that all her symptoms – including the brain fog and memory deficits – went into remission.
Schultek has since founded a research group to explore the role of infection more generally in cognitive decline. This idea would once have been considered outlandish, but interest in the brain’s microbial community is growing rapidly. It turns out our grey matter is teeming with bacteria, viruses and fungi, and a better understanding of this unexpected microbiome has enormous potential to prevent neurodegenerative diseases. It could even reverse symptoms of decline when things go awry, as Schultek found. And, most excitingly of all, some potential treatments have a proven track…
[ad_2]
Source link

[ad_1]
The abnormal tissues range from black blisters to red nodules and white-hot cysts. They grow throughout the pelvic cavity, latching on to the ovaries and peritoneum or infiltrating the bowel and bladder.
Once detected, the painful lesions of endometriosis, an inflammatory condition, can be removed. But for up to half of people who opt for this, the pain returns or persists so intensely that they need surgery again within five years. “As surgeons and doctors, we want to remove lesions. But people’s pain persists more often than we like to report,” says Amira Quevedo, an obstetrician-gynaecologist who runs endometriosis clinical trials at the University of Florida in Gainesville.
The pain experienced by people with endometriosis doesn’t reflect the number, size or type of lesions present, and varies wildly between individuals. For some, the pain worsens during their period; for others, it lasts all month long. It can manifest as searing muscle spasms, or as vaginal, bowel or bladder pain that spreads across the pelvis and beyond.
Nature Outlook: Pain
This persistence of pain after the original stimuli have subsided or been removed is a key feature of many kinds of chronic pain. In some whole-body pain conditions, such as fibromyalgia, there is no clear cause. Something has tripped the pain system into overdrive, prompting a desperate search for relief.
At least in the case of endometriosis, that relief is often found in switching diets. The foods people eat can rapidly alter the vast collection of microbes that reside in the intestine, in turn releasing chemicals that either drive or dampen pain. Observations that people with chronic pain have different mixes of microbes in their gut from other individuals have also given rise to the idea that manipulating this gut microbiome — through diet or other means — might help.
That outcome remains speculative, awaiting more clinical trials. And tinkering with the microbiome is unlikely to provide relief for everyone, especially when chronic pain becomes hardwired in the brain. But given the paucity of other options, and the potential benefits, a microbiome-focused approach is worth pursuing. It could “significantly change the way we understand, diagnose and treat chronic pain”, says Amir Minerbi, a physician at the Rambam Institute for Pain Medicine in Haifa, Israel. But researchers are wary of overstating the gut microbiome’s analgesic abilities. “We don’t want to give false hope,” Minerbi says.
The connection between the gut and chronic pain began to materialize two decades ago in studies of irritable bowel syndrome (IBS), a chronic condition more painful than its name suggests. IBS is marked by visceral pain, which emanates from organs in the abdomen and pelvis.
Despite decades of work probing the connections between gut bacteria and visceral pain in IBS, “it’s been a slow evolution” to recognize the gut microbiome’s role, says stress neurobiologist John Cryan at University College Cork in Ireland.
About 20 years ago, animal studies began to reveal that certain bacteria stimulate pain receptors on cells of the gut in a manner similar to morphine, among other mechanisms. In 2008, Cryan’s team showed that animals that had been stressed in early life by being separated from their mothers developed a whole-body syndrome of inflammation and sensitivity to visceral pain that was linked to changes in their gut bacteria1. “That was our first real strong look” at how changes in pain related to changes in the microbiome in an animal model, he says.
Steadily, researchers realized not only that the gut’s resident bacteria could induce persistent visceral pain — and, in fact, were required for normal visceral pain sensation — but also that transplanting certain microbes from one animal to another could relieve it. These findings have since been replicated in other types of pain, such as allodynia, a type of severe nerve pain stemming from very faint stimuli.
Human trials of probiotics have shown what Cryan says are “slight but significant” analgesic effects2 on visceral pain in IBS. Altogether, the evidence suggests that “the microbiome is playing a key role in pain”, he adds. It is even possible that gut bacteria influence not only how neurons transmit pain, but also how those acute pain signals turn chronic.
“It makes a lot of sense,” says Jessica Maddern, a pain researcher at the South Australian Health and Medical Research Institute in Adelaide, who lives with the visceral and chronic pelvic pain of endometriosis. The gut is studded with nerves and constantly contacting the brain through the vagus nerve and spinal cord, she says, “so it stands to reason that it could change the way you’re experiencing pain”. Diet is already known to affect mood through the ‘gut–brain axis’, but the finer details of how gut microbiota influence pain are still being worked out.

Microglia (green) can become overly reactive and affect pain signals to the brain.Credit: Gabriel Luna, NRI-UCSB/Wellcome/CC BY 4.0
Animal studies of visceral pain have identified specific chemicals, produced by gut bacteria, that can promote3 or suppress4 pain. Short-chain fatty acids, which are produced when certain bacteria digest fibre, stimulate immune cells to release pro-inflammatory factors, whereas bile acids suppress the activity of sensory nerves. The effects can be far-reaching: these metabolites can seep into the circulation through the gut lining and cross the blood–brain barrier, altering the permeability of both structures.
As a result, the gut microbiome might even influence the perception of pain. Cryan says his group’s animal studies show “very clearly” that brain regions known to be involved in the emotional and cognitive aspects of pain, such as the anterior cingulate cortex, change with alterations in the gut microbiome5. “We’re beginning to see that signals from the microbiome impinge on how visceral pain is perceived in the brain,” he says. “But we’re only scratching the surface.”
Maddern studies nerve pathways in visceral pain. Coming to this research some 20 years after her own endometriosis diagnosis, she was shocked at how little was known about pain and how to treat it. Only in the past few years have researchers begun modelling visceral pain in animal models of endometriosis, as they do for IBS. That’s because most endometriosis research has so far focused on understanding what causes the disease and the growth of lesions, rather than its pain.
Still, endometriosis — which involves interconnected and overlapping pain symptoms — could serve as a window into chronic pain more broadly. Much-improved mouse models developed by Maddern and by Kelsi Dodds, a neurophysiologist at Flinders University in Adelaide, offer fresh insights.
Maddern’s and Dodds’s work expands researchers’ understanding of the way in which pain becomes chronic, identifying how cells in the spinal cord drive a process called central sensitization. In female mice with long-standing endometriosis, the spinal cord’s resident support cells — microglia and astrocytes — amass, become overly reactive to and amplify pain signals6,7. Central pain pathways become hypersensitive to peripheral inputs, such that light touch becomes unbearable, or chronic pain persists even without any stimuli — as is common after endometriosis lesions are removed. Other researchers modelling endometriosis in mice have similarly reported swollen and therefore activated microglia in the brain8. Activated microglia are appearing in models of fibromyalgia, too9.
The anatomy of pain pathways also goes some way to explaining how the gut microbiome could affect widespread pain. Some nerve pathways innervate multiple organs in the pelvis and converge in the spinal cord, so signals from gut microbiota could very easily cross from the gut over to other pelvic organs and beyond, Dodds says.
Many women with endometriosis report finding relief by making changes to their diet. Some report that doing so completely changes their experience of the disease, says Francesca Hearn-Yeates, who is studying the impact of diet on endometriosis-associated pain at the University of Edinburgh, UK.
In an as-yet unpublished survey, she asked some 2,600 people with endometriosis about their symptoms, including bloating, cramps and pain, and what they ate. About 83% of respondents — drawn from 51 countries — said they had altered their diets. And of that subset of respondents, 63% said that these dietary modifications reduced their pain. No one diet stood out as the most effective, but going gluten-free and dairy-free often helped. “It’s not a fix-all for everyone,” she says, “but the fact that it’s benefiting so many people is really promising.”
To get at some of the mechanisms involved, she has begun an exploratory study to profile gut-bacteria metabolites in 50 people with suspected endometriosis who are awaiting diagnostic surgery. Multiple studies have shown that people with endometriosis have altered gut microbiomes, yet few have examined how that microbial community functions as a collective10. And relating people’s metabolite profiles to their diets and pain, as she plans to do, is new territory. “There’s clearly this really intricate interaction between the gut and the brain,” she says. But the task that looms ahead is to pin down how specific bacteria influence pain.
Research in animals is looking to antibiotics as a way to manipulate the gut microbiome — and by extension, chronic pain. Two studies have found that metronidazole, an antibiotic used to treat gastrointestinal and reproductive tract infections, can shrink endometriosis lesions in mice11, and even stop them forming12.
This makes sense: people with endometriosis have an abundance of anaerobic bacteria sensitive to metronidazole in their gut. However, neither study looked at pain. So Quevedo and her colleagues at the University of Louisville Hospital in Kentucky are investigating whether administering endometriosis after surgery could reduce pain.
Starting in 2020, 72 people with endometriosis randomly received either low-dose metronidazole or a placebo for two weeks. The two groups showed no differences in pain six weeks after surgery13, but Quevedo remains optimistic. The trial runs until 2027, and participants will report their symptoms six months after surgery and annually for five years, a timeline more relevant to chronic pain.
However, Quevedo admits that antibiotics alone might not be enough to quell persistent pain. They could help to ‘reset’ the gut microbiome by removing problematic bacteria. But achieving a sustained benefit will probably require probiotics — which seed the gut with beneficial bacteria — along with dietary changes promoting microbial strains linked to reduced pain.
Two small randomized trials suggest that taking daily probiotics containing selected Lactobacillus strains can reduce painful periods in people with endometriosis10, but the relief seems short-lived. This transient effect probably reflects the complexity of human pain experiences compared with mouse models, Quevedo says; after enduring endometriosis for years, most individuals have persistent pain that doesn’t budge.
If clinician-scientists are to find other ways to ease pain, Quevedo says, they need to differentiate between chronic pain types and between various forms of endometriosis. This stratification of clinical subtypes is often missing from microbiome studies that lump patients together, but is necessary to work out which therapies alleviate whose pain. The same is true of chronic pain generally; pain is deeply personal and what eases one person’s discomfort might do little for someone else. “We know that one treatment is not going to help everybody,” Quevedo says.
The role of the gut microbiome is becoming clearer in chronic pain conditions that are not visceral in origin. The foremost example is fibromyalgia, a pain disorder that typifies central sensitization — it causes widespread pain in joints, muscles and tendons, and shattering fatigue, but is often misdiagnosed.
Minerbi’s research suggests that the gut microbiome could not only help to ease fibromyalgia pain, but also aid in diagnosing it and other chronic pain conditions. In 2019, his team found that people with fibromyalgia have altered gut microbiomes, and that those slight imbalances correlated with pain and fatigue — and not with diet, medications or other environmental factors14. According to Minerbi, it was the first demonstration in humans that the gut microbiome might modulate widespread, non-visceral chronic pain.
In a 2023 study, the team found that people with fibromyalgia also had lower levels of specific bile acids in their blood compared with healthy controls15. These secondary bile acids are produced by gut bacteria that people with fibromyalgia tend to lack. In fact, the lower the level of bile acids they had circulating, the more intense pain they reported — possibly because some of these acids bind to neurons in the spinal cord that suppress pain. Without them, pain might flare unchecked. The study suggests that restoring the levels of these bile acids could help to reduce fibromyalgia pain.

Amir Minerbi prepares gut microbiome samples for analysis.Credit: Rambam Health Campus.
Minerbi and his team have tried transplanting faecal matter from healthy donors into 14 women with fibromyalgia to address such gut-microbiome imbalances. After five fortnightly treatments, 12 of the volunteers in this pilot study, which has not been peer-reviewed, reported less severe pain than before9. A randomized, placebo-controlled trial is next.
Minerbi’s group is also developing a machine-learning algorithm to relate chronic pain to gut microbiome profiles and other blood markers. So far, the tool differentiates only between people with fibromyalgia and those who do not have chronic pain14,15. The next step, Minerbi says, is to see “whether we can take someone with chronic pain and say what type of chronic pain they have, which is really the clinical question”. To that end, researchers are investigating the gut microbiome in various chronic pain conditions, hoping to find commonalities and distinct changes between them, as one 2024 study has done16.
This work has just begun. So far, most human studies have involved broad-stroke characterizations of the gut microbiomes of people with chronic pain conditions and those without. These differences — some subtle, others striking — implicate the gut microbiome in a gamut of chronic conditions, from inflammatory arthritic pain and migraine headaches to nerve-injury pain. Whether these observed differences are an underlying cause of those conditions or a knock-on effect, however, remains unclear.
Most studies capture only a snapshot of the gut microbiome. Therefore, Cryan says, large, longitudinal studies are needed to track microbiome changes in response to symptoms and treatment. His research in animals shows that early life stress affects the gut microbiome in ways that lead to persistent visceral pain in adulthood, even though the microbiome itself recovers17. “When you’re looking at pain, the microbiome may not be a reflection of that pathology; it might be something that happened way earlier that affected pain processes,” he says.

More from Nature Outlooks
Still, Cryan thinks that modifying the gut microbiota could help to relieve chronic pain. He cites animal studies showing that specific probiotic strains can reverse well-established pain, even when given in adulthood18. That apparent plasticity offers some hope, but he says it’s essential for researchers to investigate which strains of bacteria relieve chronic pain in humans — and how long that effect lasts.
Despite these challenges, researchers are keeping an open mind that the gut microbiome could help to ease chronic pain. So immense is the burden, Maddern says, that “everything is worth trying at this point”.
[ad_2]
Source link

[ad_1]

Fermented foods such as kimchi contain microbes that are also found in the human microbiome.Credit: Chung Sung-Jun/Getty
You are what you eat — at least when it comes to the microbiome. A catalogue of microorganisms from more than 2,500 cheeses, meats and other foods suggests that a small portion of each person’s microbiome comes from the food they eat. The study1 is the largest-ever compendium of bacteria, fungi and other microbes found in foods.
Some microbes are an essential ingredient of fermented foods — from salami to sauerkraut and kimchi to kefir. Other microorganisms in fermented and unfermented foods could be important to their taste and other properties such as how quickly they spoil, says microbiologist Nicola Segata at the University of Trento, Italy, who co-led the work, published in Cell on 29 August.
Segata and his colleagues sequenced microbial DNA from nearly 2,000 foods and collated these data with almost 600 existing food microbiomes. Most of the foods were fermented — Segata was sure to include samples of Gorgonzola cheese, one of his favourites — but the study also included fresh meat, fish, fruit and vegetables.
Similar foods tended to harbour similar microbes, although a closer look revealed some intriguing patterns. Lactic acid-making bacteria including Lactobacillus were especially prevalent in dairy products, but the composition varied. Dutch blue cheese harboured different Lactobacillus species from Italian fontina and mozzarella, for instance. Microbes from coffee, kombucha and pu’er — a fermented tea from Yunnan, China — resembled those in alcoholic beverages.
Nearly every microbiome study uncovers organisms that have never been seen before, and this one was no different. About half the microbes the researchers identified were new. Pulque — a sour agave wine drunk in Mexico — was especially rich in this microbial dark matter, as were African palm wine and cheese brine.
When the researchers compared the food microbiomes with thousands of microbiomes from human guts and mouths, they found a degree of overlap. About 3% of the microbe species in adults’ guts, 8% of children’s and more than 50% of newborns’ were also found in food. This doesn’t necessarily mean that these microbes all came from foods people ate, Segata says: the overlap could also reflect instances in the past when food microbes became established in peoples’ guts and began circulating between humans. The food microbes in newborns’ microbiomes tended to be associated with dairy but are also found in breast milk.
None of these findings is especially surprising, says Benjamin Wolfe, a microbiologist at Tufts University in Medford, Massachusetts. But the study lays the groundwork for detailed research into why various microbes — and communities of microbes — are in particular foods. He’s also intrigued by all the unknown microbes in what we eat. Mining these, Wolfe says, could lead to new kinds of food with novel properties.
[ad_2]
Source link
[ad_1]
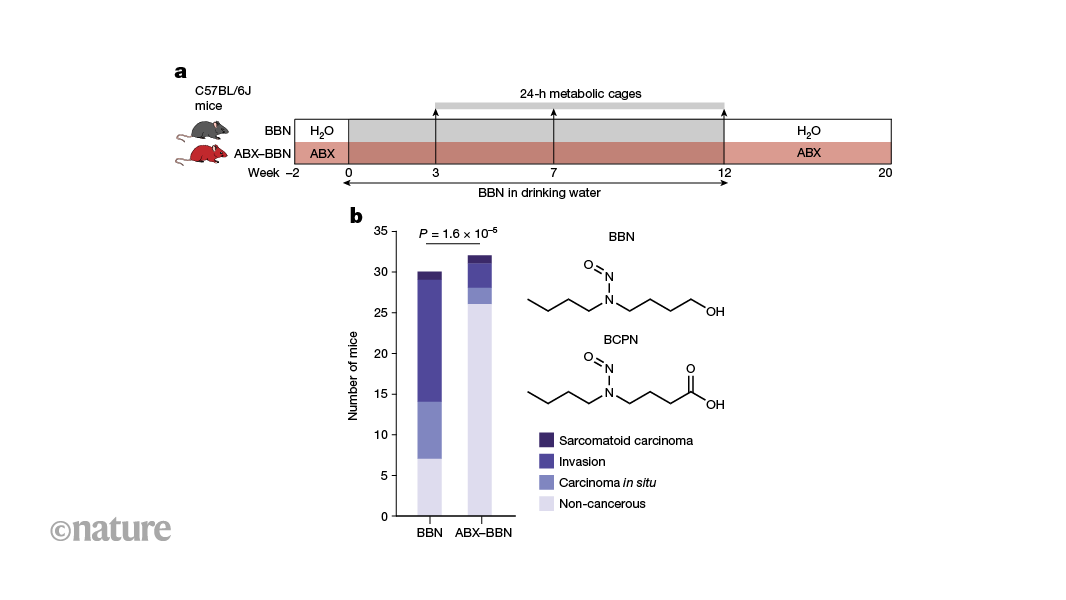
Nature, Published online: 28 August 2024; doi:10.1038/d41586-024-02740-8
Bacteria in the gut can affect the chemically induced growth of tumours in other parts of the body. Certain gut bacteria convert nitrosamine compounds, which are found in cigarette smoke and processed food, into metabolites that trigger the growth of tumours in the bladders of mice.
[ad_2]
Source link

[ad_1]

Crohn’s disease, a form of inflammatory bowel disease, often affects the intestines (artist’s illustration).Credit: Sebastian Kaulitzki/Science Photo Library
When geneticist James Lee and his colleagues published a paper in June linking a gene to inflammatory bowel disease (IBD), he didn’t expect the public to take much notice. Things did not go as planned.
“I got inundated,” he says.
By the end, Lee did more than 25 interviews for radio and print outlets around the world and received hundreds of e-mails from people with IBD. “It’s a testament to how common inflammatory bowel disease is,” says Lee, who works at the Francis Crick Institute in London. “And also a testament to how desperate people are for better treatments.”
Lee’s paper, published in Nature1, is one of several recent reports offering hope that people with IBD could one day have better treatment options tailored to their disease. Lee and his colleagues found that changes in the activity of a gene that is important in the immune system could contribute to some cases of the disease. Another study found that some people with IBD make antibodies that disable a pivotal anti-inflammatory protein2 and a third study tracked how populations of gut bacteria adapt to an inflamed environment3.
The papers look at IBD from different angles, but together offer a glimpse into the ways that physicians might one day be able to better match people with IBD to appropriate treatments, says David Artis, an immunologist at Weill Cornell Medicine in New York City. “Not every inflammatory bowel disease patient who walks in the door is the same,” he says. “If we can map that difference to some extent, I think we’re going to be able to better treat those people.”
IBD is a painful condition that causes chronic inflammation of the digestive tract. Two of the most common forms of IBD are ulcerative colitis and Crohn’s disease. Both can cause diarrhoea, anaemia and abdominal cramping.
Like many autoimmune disorders, IBD has an aetiology that is murky and complex, with contributions from both genetics and the environment. What is clear is that incidence of the disease is rising in many regions of the world4.
Over the past decade, researchers have amassed a lengthy list of genetic variations that are linked to IBD. But Lee and his colleagues decided to examine a region of the genome where few geneticists had bothered to look: a “gene desert”, says Lee, so named because it is devoid of any recognizable genes. “We didn’t know what we were going to find,” he says. “And we ended up finding a master regulator of inflammatory responses.”

Mucus-producing cells (pink; artificially coloured) stud the intestines of a person with ulcerative colitis, a common form of inflammatory bowel disease.Credit: Steve Gschmeissner/Science Photo Library
This master regulator is a stretch of DNA that controls the activity of a gene called ETS2, which is located far away from the gene desert. High levels of ETS2 activity, the team found, boost the ability of immune cells called macrophages to promote inflammation.
The finding also indicated that a class of cancer drugs called MEK inhibitors might prevent the activation of ETS2. The team found that these drugs could block the effects of the ETS2 protein, including the release of inflammation-promoting molecules, in cells grown in the laboratory. But MEK inhibitors can become toxic to other cells if given over the long term, says Lee, and so the team is developing ways to deliver the inhibitors only to macrophages before testing the approach in people with IBD.
Another study has found a select group of people with IBD who might have a new therapeutic option in the near future. Paediatric immunologist Sophie Hambleton at Newcastle University in Newcastle upon Tyne, UK, and her colleagues analysed samples from two children with IBD. The scientists discovered that the children were producing antibodies that block the activity of a protein called IL-102. This protein has anti-inflammatory effects in the gut.
But the children’s antibodies meant that IL-10 was unable to dampen inflammation in their intestines, leading to IBD, the researchers reported in July in the New England Journal of Medicine. Once the link between IL-10 and their disease was identified, one of the children was treated with therapies to deplete the antibodies, easing their symptoms.
It’s unclear how many people with IBD make antibodies against their own IL-10, says Hambleton. But when the team looked at a sample of adults with IBD, they found “a clear minority” who also produced the antibodies. “We are very confident that this is going to be a contributory mechanism in more patients,” she says.
In addition to genetics and immune cells, microorganisms are thought to play a part in IBD. In the third study, Christopher Smillie, who studies the human microbiome at Harvard Medical School in Boston, Massachusetts, and his colleagues, looked at how chronic inflammation shapes evolution of the microorganisms living in the digestive track3.

Gut feeling yields evidence of microbial involvement in autoimmunity
They identified 140,000 bacterial strains in stool samples from people with and without IBD. Hundreds of these strains were associated with IBD, and many appear to have adapted to living in inflamed tissue. Among those, several could be used to predict disease severity: for example, the abundance of some strains of Eggerthella lenta declined as the levels of a protein associated with inflammation rose. The results were published in Cell Host & Microbe in July.
Ultimately, Smillie hopes that characterization of these microorganisms will lead to ways to monitor disease progression, and to sort people with IBD into groups on the basis of how likely they are to respond to potential therapies.
Each of these studies could contribute to that goal, but the work is still preliminary, says Gabriel Nuñez, an immunologist at the University of Michigan Medical School in Ann Arbor. For example, the microbial study does not show that any of these organisms contribute to disease, he notes. And it is unclear what proportion of people with IBD have altered ETS2 activity or make autoantibodies against IL-10. “Perhaps these are rare patients, and only a handful in the world will benefit,” he says.
Nevertheless, if only a handful of people find relief because of these results, that will be progress, he adds. “Even if you cure only one patient, it’s important for that person and their family.”
[ad_2]
Source link
[ad_1]

The vagus nerve connects the brain region called the amygdala (red; artist’s illustration) to nerves for the Brunner’s glands in the gut.Credit: Sebastian Kaulitzki/Science Photo Library
Stress can make people feel sick, and bacteria in the gut might be to blame, according to a study1 in mice. The research suggests that a stressed brain directly shuts down specific glands in the gut, affecting gut bacteria and the body’s broader immune system.
The study “is a technical tour de force”, says neuroscientist John Cryan at University College Cork in Ireland, who reviewed the study. Most work on the gut–brain connection has focused on how bacteria affect the brain, so Cryan welcomes research into how psychological states can exert ‘top-down’ control of bacteria. “It’s a really cool part of the puzzle”, he says. The research was published on 8 August in Cell.
Researchers have long known that the gut and brain ‘talk’ to each other. Under stress, the brain spurs the release of hormones that can trigger gut conditions such as inflammatory bowel disease. And certain bacteria in the gut can release chemical signals that affect the brain and behaviour.

Your brain could be controlling how sick you get — and how you recover
But the neural communication pathways are less understood. To find out more, neuroscientist Ivan de Araujo at the Max Planck Institute for Biological Cybernetics in Tübingen, Germany, and his colleagues focused on small organs called Brunner’s glands that are found in the walls of the small intestine. Little is known about these glands, other than that they produce mucus and contain numerous neurons.
De Araujo’s team found that removing the Brunner’s glands of mice made the animals more susceptible to infection. It also raised markers of inflammation, a flood of immune chemicals and cells that can damage tissues. The team saw a similar effect in humans: people who’d had tumours removed from the part of the gut containing Brunner’s glands had higher levels of white blood cells — a marker of inflammation — than people who’d had tumours removed from other areas.
Closer analysis showed that removing the Brunner’s glands from mice eliminates bacteria in the Lactobacillus genus, which live in the small intestine. In a healthy gastrointestinal tract, Lactobacilli stimulate production of proteins that act as grout between the cells lining the gut, keeping most of the gut’s contents inside while allowing certain nutrients to enter the bloodstream. But when Lactobacilli are gone, the gut becomes ‘leaky’ and “things that shouldn’t cross into the blood do so”, de Araujo says. The immune system attacks these foreign molecules, causing the inflammation and illness seen in mice without Brunner’s glands.
The researchers then examined the glands’ neurons. They found that the neurons connect to fibres in the vagus nerve, a communications pathway between the gut and the brain. These fibres run directly to the brain’s amygdala, which is involved in emotion and stress response.
Placing mice with intact Brunner’s glands under chronic stress had the same effect as removing the glands: Lactobacillus levels fell, and inflammation increased. This suggested that stress had shut down the Brunner’s glands.
Asya Rolls, a neuroimmunologist at the Technion — Israel Institute of Technology in Haifa, is impressed by the direct line between the brain, Brunner’s glands, bacteria and immune system. “The specificity of the connection is amazing,” she says. But she cautions that the pathways in mice might not be identical to those in humans.
“This paper is pretty inspiring,” says Christoph Thaiss, a microbiologist and neuroscientist at the University of Pennsylvania in Philadelphia. Understanding the specific pathways that connect the brain and gut, he says, could help researchers to study questions such as why some people are more resilient to stress than others.
De Araujo says the study could have implications for treating stress-related disorders such as inflammatory bowel disease. His group is now studying whether chronic stress affects this pathway in infants, who receive their Lactobacillus through breast milk. “We are excited about the idea that these glands are important for normal development and immune function early in life,” de Araujo says.
[ad_2]
Source link