[ad_1]
Nature, Published online: 26 September 2024; doi:10.1038/d41586-024-02948-8
Genetic evidence kick-starts research into the involvement of microglia cells and the innate immune system in the development of the disease.
[ad_2]
Source link
[ad_1]
Nature, Published online: 26 September 2024; doi:10.1038/d41586-024-02948-8
Genetic evidence kick-starts research into the involvement of microglia cells and the innate immune system in the development of the disease.
[ad_2]
Source link
[ad_1]
Palmqvist, S. et al. JAMA 324, 772–781 (2020).
Jack, C. R. Jr et al. Alzheimer’s Dement. 14, 535–562 (2018).
Janelidze, S. et al. Nature Med. 26, 379–386 (2020).
Thijssen, E. H. et al. Nature Med. 26, 387–397 (2020).
Janelidze, S. et al. Nature Commun. 11, 1683 (2020).
Mattsson-Carlgren, N. et al. Brain 143, 3234–3241 (2020).
Mielke, M. M. et al. Nature Med. 28, 1398–1405 (2022).
Barthélemy, N. R. et al. Nature Med. 30, 1085–1095 (2024).
[ad_2]
Source link
[ad_1]
Nature, Published online: 26 September 2024; doi:10.1038/d41586-024-02952-y
The arrival of the first disease-modifying therapy for Alzheimer’s was significant, but it was not met with the joy that might have been expected.
[ad_2]
Source link
[ad_1]
Nature, Published online: 11 September 2024; doi:10.1038/d41586-024-02862-z
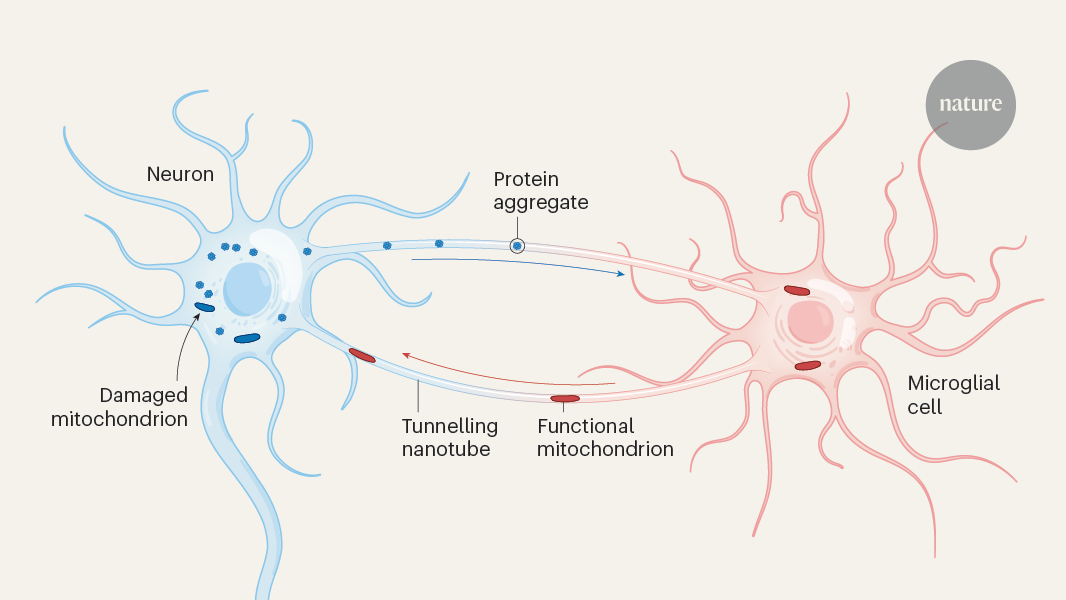
Tiny cellular tubes between neurons and brain cells called microglia serve as conduits for the export of toxic protein aggregates from neurons and the import of healthy organelles, keeping neurodegeneration at bay.
[ad_2]
Source link

[ad_1]

A functional magnetic resonance imaging scan (artificially coloured) of a human brain. Researchers used data from such scans to measure an aspect of brain ageing.Credit: Mark and Mary Stevens Neuroimaging and Informatics Institute/Science Photo Library
A newly devised ‘brain clock’ can determine whether a person’s brain is ageing faster than their chronological age would suggest1. Brains age faster in women, countries with more inequality and Latin American countries, the clock indicates.
“The way your brain ages, it’s not just about years. It’s about where you live, what you do, your socioeconomic level, the level of pollution you have in your environment,” says Agustín Ibáñez, the study’s lead author and a neuroscientist at Adolfo Ibáñez University in Santiago. “Any country that wants to invest in the brain health of the people, they need to address structural inequalities.”
The work is “truly impressive”, says neuroscientist Vladimir Hachinski at Western University in London, Canada, who was not involved in the study. It was published 26 August in Nature Medicine.
The researchers looked at brain ageing by assessing a complex form of functional connectivity, a measure of the extent to which brain regions are interacting with one another. Functional connectivity generally declines with age.
The authors drew on data from 15 countries: 7 (Mexico, Cuba, Colombia, Peru, Brazil, Chile and Argentina) that are in Latin America or the Caribbean and 8 (China, Japan, the United States, Italy, Greece, Turkey, the United Kingdom and Ireland) that are not. Of the 5,306 participants, some were healthy, some had Alzheimer’s disease or another form of dementia and some had mild cognitive impairment, a precursor to dementia.
Five ways the brain can age: 50,000 scans reveal possible patterns of damage
The researchers measured participants’ resting brain activity — that when they were doing nothing in particular — using either functional magnetic resonance imaging (fMRI) or electroencephalography (EEG). The first technique measures blood flow in the brain, and the second measures brain-wave activity.
The authors computed the functional connectivity of each person’s brain and input those data into two deep-learning models trained to predict brain age, one for fMRI data and one for EEG data. They could then calculate each person’s ‘brain-age gap’ — the difference between their chronological age and their brain age estimated from functional connectivity. Having a brain age gap of ten years, for example, would mean that your brain connectivity is roughly the same as that of someone ten years older than you.
The models showed that people with Alzheimer’s or another type of dementia had larger brain-age gaps than both those with mild cognitive impairment and healthy controls.
Participants from Latin American or the Caribbean had larger brain-age gaps, on average, than did those from other regions. Latin America is one of the most unequal region in the world, says Ibáñez, and he thinks that this is why the brains of people from that region aged faster. Structural socioeconomic inequality, exposure to air pollution and health disparities were linked to larger brain-age gaps, especially in people from Latin America.
Moreover, women living in countries with high gender inequality — particularly those in Latin America and the Caribbean — tended to have larger brain-age gaps than did men in those countries.
Simply quantifying brain ageing in a sample this geographically diverse is a phenomenal achievement, says Hachinski. He thinks the conclusion that brain-age gaps vary is solid, but he cautions that functional connectivity is only one way of measuring the health of the brain, and that someone could have a lot of brain connectivity while having, for example, poor mental health due to conditions such as depression or anxiety. Neuroscience is “not good at measuring gestalts”, he says.
Blood tests could soon predict your risk of Alzheimer’s
One possible source of inconsistency in the data is the range of fMRI machines and EEGs — spread across 15 nations — that supplied the brain scans. For example, lower-income nations might have had older equipment that generated lower-quality data than those from higher-income nations. But Ibáñez found no association between lower data quality and either a larger brain-age gap or higher structural inequality.
Now, Ibáñez’s team is investigating whether brain-age gaps are linked to national income by comparing brain-age gaps in groups from Asian nations and the United States, and adding data from ‘epigenetic’ clocks that measure biological age by examining chemical modifications on DNA. Eventually, Ibáñez hopes that the data will contribute to personalized-medicine approaches that are grounded in the full biological diversity of people’s brains around the world.
“We need to understand this diversity,” says Ibañez. “We cannot create a truly global science of dementia without addressing this.”
[ad_2]
Source link
[ad_1]

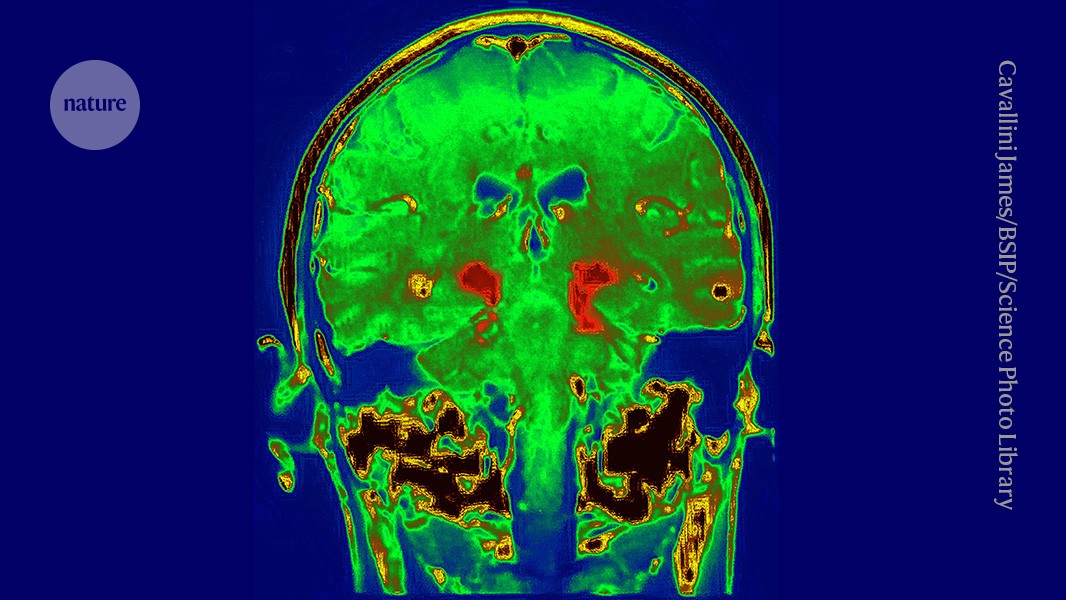
Superimposed scans show the brain of a person with Alzheimer’s disease. Credit: Zephyr/Science Photo Library
A drug for Alzheimer’s disease has won unanimous approval from independent scientists advising the US Food and Drug Administration (FDA), bringing the treatment closer to use in the clinic.
The drug, donanemab, would be the second on the US market that slows the cognitive decline caused by the disease. But donanemab’s effects are modest, it does not reverse symptoms and the FDA might limit who can take it.
At a 10 June meeting at the FDA’s headquarters in Silver Spring, Maryland, all 11 members of an FDA independent scientific advisory committee voted that donanemab, made by Eli Lilly, based in Indianapolis, Indiana, is effective at treating Alzheimer’s, at least in people at early stages of the disease, and that its benefits outweigh its risks.
The advisory meeting itself was a surprise to many observers, who had expected the FDA to quickly approve donanemab without convening an advisory committee. Instead the FDA delayed its decision until after a public meeting could be held because of questions about the drug’s efficacy in people with certain markers of Alzheimer’s disease. But in the end, “it was a very positive meeting,” says neurologist David Knopman of the Mayo Clinic in Rochester, Minnesota, who was not on the committee. “It would have been difficult for someone to object based on the standard of the data.”
Donanemab is an antibody that attacks amyloid, a sticky protein that accumulates in the brains of people with Alzheimer’s. In data submitted to the FDA, Lilly reported that the 860 trial participants who received donanemab lost their cognitive abilities more slowly over the course of 18 months than those who received a placebo. The drug, however, did not reverse the disease. Research1,2 shows that donanemab slows symptoms roughly as well as rival drug lecanemab, which also attacks amyloid. It is made by bio-pharmaceutical companies Eisai in Tokyo and Biogen in Cambridge, Massachusetts.
More Alzheimer’s drugs head for FDA review: what scientists are watching
Unlike previous trials of monoclonal antibodies, Lilly only tested people whose brains contained both amyloid and another protein called tau that is associated with cognitive decline. Donanemab seemed more effective in people who had low to moderate tau levels at the start of the trial than in people who had high levels. But the FDA pointed out that the disease might have progressed more slowly in the lower-tau group than it in those with higher tau.
At the meeting, advisory committee members were broadly supportive of the drug. Some noted, however, that Lilly has little evidence that it works in people with no to very little tau. But the committee decided against restricting the drug’s usage on the basis of tau levels, because screening for tau is complex and costly. A screening requirement would prevent an unacceptably high number of people from accessing the drug, the committee decided.
The panel members also expressed some concerns about a condition called amyloid-related imaging abnormalities (ARIA) that can cause brain bleeding and seizures. ARIA, which can be fatal, is thought to occur when the antibodies weaken blood vessels in the brain. Lilly recorded more ARIA-related deaths among people who received the drug than in the placebo group. Lecanemab also has been linked to ARIA, but the increased risk seems to be much higher with donanemab3.
The approval is a bright spot for amyloid-targeting Alzheimer’s drugs after numerous controversies. The FDA approved the first such drug, Biogen and Eisai’s aducanumab, in 2021 over the objections of its advisory committee, leading three committee members to resign in protest. A US Congressional investigation found that that the FDA had improperly guided the manufacturers through the approval process. Many insurers were unconvinced by the drug’s efficacy, and most refused to cover it. Biogen stopped making the drug this year. And three people died from ARIA-related conditions during clinical trials of lecanumab.
Landmark Alzheimer’s drug approval confounds research community
The donanemab committee said that more research is still needed on the drug, including how long people should take it, and its efficacy in people with different levels of tau. Knopman says that it remains to be seen whether the drug’s modest effect will persist for years.
The committee also recommended more research on the drug’s efficacy in people of colour — more than 90% of Lilly’s trial participants were white — and people with Down’s syndrome or genetic mutations that make them more prone to ARIA. Committee member Kathleen Poston, a neurologist at Stanford University Medical Center in California, says scientists need to obtain these data “to make sure that these encouraging findings can be extrapolated to everyone with Alzheimer’s disease.” The FDA’s final decision is expected later this year.
[ad_2]
Source link

[ad_1]
A study by the University of the Basque Country (UPV/EHU) and Biobizkaia proposes using an available, simple, non-invasive tool to monitor this neurodegeneration.
Although there are still some aspects pending confirmation for its use in the clinical setting, and its resolution needs to be improved slightly, a study by the UPV/EHU and Biobizkaia has shown that a method routinely used to carry out ophthalmological tests can also be used to monitor the neurodegeneration that occurs in Parkinson’s patients. In the course of the research it was found that the neurodegeneration of the retina probably precedes cognitive impairment.
When Parkinson’s or another neurodegenerative disease is diagnosed, patients always ask: “And now what? What will happen? What can be expected from the disease?” For neurologists, however, it is not possible to answer these questions precisely, as “the evolution of patients tends to be very varied: some experience no change over the years, while others end up with dementia or in a wheelchair”, explained Ane Murueta-Goyena, researcher in the UPV/EHU’s department of Neurosciences.
Today, identifying Parkinson’s patients at risk of cognitive impairment poses a major challenge, yet this is necessary when it comes to providing more effective clinical treatments and stepping up clinical trials. In fact, Dr. Ane Murueta-Goyena, in collaboration with Biobizkaia’s research staff, wanted to see “whether the visual system can enable this deterioration to be predicted, in other words, what future the patient can expect within a few years”. The thickness of the retina was used for this purpose.
The retina is a membrane located at the back of the eyeball, it is related to the nervous system and comprises several layers. During the study, a cohort of Parkinson’s patients had the thickness of the innermost layer of their retinas measured using optical coherence tomography. This type of tomography is a routinely used instrument in ophthalmological tests, as it allows high-resolution, repeatable and accurate measurements to be made. So the evolution of this retinal layer was analysed and compared in people with and without Parkinson’s disease over the 2015-2021 period. The results of the analysis of the images of the retinal layers of Parkinson’s patients were also confirmed in a UK hospital.
The results showed that the retinal layer is noticeably thinner in Parkinson’s patients. It was also observed that “during the initial phases of the disease it is in the retina where the greatest neurodegeneration is detected, and, from a given moment onwards, when the layer is already very thin, a kind of stabilising of the neurodegeneration process takes place. Retinal thinning and cognitive impairment do not occur simultaneously. The initial changes in the retina are more evident and then, over the years, patients are observed to worsen clinically in both cognitive and motor terms”, explained Murueta-Goya. In other words, the slower retinal layer thickness loss is associated with faster cognitive decline; this slowness is linked to greater severity of the disease”.
The researcher has attached great importance to the results: “We have obtained information on the progression of the disease, and the tool we are proposing is non-invasive and available at all hospitals.” The results need to be validated internationally and “by slightly improving the resolution of the technology, we will be closer to validating the method for monitoring the neurodegeneration that takes place in Parkinson’s disease”. The researcher also revealed that they are continuing the research on another cohort of patients and that funding is the key.
Source:
Journal reference:
Murueta-Goyena, A., et al. (2024). Association of retinal neurodegeneration with the progression of cognitive decline in Parkinson’s disease. Npj Parkinson’s Disease. doi.org/10.1038/s41531-024-00637-x.
[ad_2]
Source link

[ad_1]
For the roughly 1.5 million Americans per year who survive a traumatic brain injury, health outcomes vary widely. Not only can these injuries lead to a loss of coordination, depression, impulsivity, and difficulty concentrating, but they come with an amplified risk for developing dementia in the future.
The glaring absence of treatments for such a widespread condition drove a team of scientists at Gladstone Institutes to uncover, on a molecular level, how traumatic brain injuries trigger neurodegeneration-;and just as importantly, how to target that process to prevent long-term damage.
“We set out to address the fundamental question of exactly what happens in the brain after injury to ignite the damaging process that destroys neurons,” says Jae Kyu Ryu, PhD, a scientific program leader in the lab of Katerina Akassoglou, PhD at Gladstone Institutes.
Most traumatic brain injuries come as a result of falls, car crashes, or violent assaults, according to the Centers for Disease Control, but many also stem from sports accidents or certain military operations such as explosions. In each case, the external force is strong enough to move the brain within the skull, causing a significant breakdown in the blood-brain barrier and allowing blood to move in.
“We knew that a specific blood protein, fibrin, was present in the brain after traumatic brain injury, but we didn’t know until now that it plays a causative role in brain damage after injury,” says Ryu, who led the study that appears in the Journal of Neuroinflammation.
Ryu and others in Akassoglou’s lab have long investigated how blood that leaks into the brain triggers neurologic diseases, essentially by hijacking the brain’s immune system and setting off a cascade of harmful, often-irreversible effects. Fibrin, a protein that normally helps blood coagulate, is the culprit.
Across many neurological diseases, toxic immune responses in the brain are triggered by blood leaks and drive neurodegeneration. Neutralizing the toxic immune responses in the brain paves the way to new therapies for neurological diseases.”
Katerina Akassoglou, senior investigator at Gladstone and director of the Center for Neurovascular Brain Immunology at Gladstone and UCSF
In diseases such as Alzheimer’s and multiple sclerosis, abnormal leaks in the protective blood-brain barrier allow fibrin to seep into areas responsible for cognitive and motor functions causing neurodegeneration. But in this case, the traumatic brain injury itself causes the blood to leak into the brain. The new study showed, for the first time, that fibrin is responsible for turning good immune cells bad, causing dangerous inflammation and unleashing toxins that kill neurons.
The Gladstone team used state-of-the-art imaging technology to study mouse brains, as well as brains from people who experienced a traumatic brain injury. They also produced three-dimensional imaging of a whole intact mouse brain, showing blood-brain barrier leaks and abundant fibrin in traumatic brain injury. In both mouse and human brains, fibrin was present together with activated immune cells.
“It became clear that fibrin is activating these immune cells,” Ryu says. “We realized that we can prevent the toxic effects if we could block fibrin, but we had to do it in a precise way.”
The team leveraged genetic tools with a specific mutation in fibrin that can block it from activating immune cells without affecting the protein’s beneficial blood-clotting abilities. This is especially critical for traumatic brain injuries, as excessive bleeding into the brain has been known to occur among patients who were taking anticoagulant medications before their injury.
Akassoglou’s lab previously developed a drug, a therapeutic monoclonal antibody, that acts only on fibrin’s inflammatory properties, without adverse effects on blood coagulation. This fibrin-targeting immunotherapy protects from multiple sclerosis and Alzheimer’s disease in mice. A humanized version of this first-in-class fibrin immunotherapy is already in Phase 1 safety clinical trials by Therini Bio.
“It’s exciting to have a therapeutic option to neutralize blood toxicity in neurologic diseases,” Ryu says. “Future studies are needed to test the effects of the fibrin immunotherapy in traumatic brain injury.”
“This study identifies a potential new strategy to diminish the devastating impacts of brain injuries,” says Lennart Mucke, MD, director of the Gladstone Institute of Neurological Disease. “Brain injuries can have profound effects on a person’s cognitive abilities, emotional health, and motor skills, touching every part of their life. It will be interesting to explore whether blocking the disease-promoting effects of fibrin can improve the outcome of brain surgeries and reduce disability when implemented after traumatic brain injuries have occurred.”
Source:
Journal reference:
Dean, T., et al. (2024). Fibrin promotes oxidative stress and neuronal loss in traumatic brain injury via innate immune activation. Journal of Neuroinflammation. doi.org/10.1186/s12974-024-03092-w.
[ad_2]
Source link
[ad_1]
A diabetes drug related to the latest generation of obesity drugs can slow the development of the symptoms of Parkinson’s disease, a clinical trial suggests1. Participants who took the drug, called lixisenatide, for 12 months showed no worsening of their symptoms — a gain in a condition marked by progressive loss of motor control.
Further work is needed to control side effects and determine the best dose, but researchers say that the trial marks another promising step in the decades-long effort to tackle the common and debilitating disorder.
“This is the first large-scale, multicentre clinical trial to provide the signs of efficacy that have been sought for so many years,” says Olivier Rascol, a Parkinson’s researcher at Toulouse University Hospital in France, who led the study.
Lixisenatide is a glucagon-like peptide-1 (GLP-1) receptor agonist, making it part of a large family of similar compounds used to treat diabetes and, more recently, obesity. (The weight-loss drug semaglutide, sold under the brand name Wegovy, is a GLP-1 compound.)
Many studies have shown a link between diabetes and Parkinson’s2. People with diabetes are around 40% more likely to develop Parkinson’s. And people who have both Parkinson’s and diabetes often see more rapid progression of symptoms than do those who have only Parkinson’s.
Animal studies3 have suggested that some GLP-1 drugs, which influence levels of insulin and glucose, can slow the symptoms of Parkinson’s. Smaller trials, published in 20134 and 20175, suggested that the GLP-1 molecule exenatide, another diabetes drug, could do the same in people.
In the latest, larger study, the French researchers investigated lixisenatide in 156 people with mild to moderate Parkinson’s symptoms, all of whom were already taking the standard Parkinson’s drug levodopa or other drugs. Half got the GLP-1 drug for a year and the others received a placebo.
After 12 months, those in the control group showed a worsening of their symptoms. Specifically, their score had increased by three points on a scale used to assess the severity of Parkinson’s that measures how well people can perform tasks including speaking, eating and walking.
Those taking the drug had no change in their scores on this scale. But the treatment did induce side effects. Nausea occurred in nearly half, and vomiting in 13%, of people on the medication. The results are published in The New England Journal of Medicine.
David Standaert, a neurologist at the University of Alabama at Birmingham, who was not involved in the trial, says it’s important to know whether the effect will last beyond a year.
“We’re all cautious. There’s a long history of trying different things in Parkinson’s that ultimately didn’t work,” he says. A difference of three points in the rating score is a small change — one that many people with Parkinson’s would struggle to notice, he says. “What happens at 5 years? Is it 15 points then, or is it still 3? If it’s still 3, then this is not worth it.”
Lixisenatide as a diabetes treatment was pulled from the US market last year by its Paris-based manufacturer Sanofi for commercial reasons. But Standaert says that this would not have affected development of a possible treatment for Parkinson’s, because other GLP-1 drugs are available.
Obesity drugs have another superpower: taming inflammation
“I view this as a study of the class. I don’t know if this particular one is the right answer,” he says. Newer GLP-1 drugs (lixisenatide was developed in the 2000s) could offer fewer and milder side effects or work at lower doses, he adds.
Another question that needs further consideration is just how some GLP-1 drugs might protect against Parkinson’s. The compounds are known to reduce inflammation, which has led some researchers to suggest that they prevent the steady loss of dopamine-producing neurons that drives the condition. That would offer a significant benefit over existing treatments such as levodopa, which mask the symptoms but don’t address the underlying cause. But this trial and others haven’t assessed neuron loss.
Researchers are now waiting for the results of a large clinical trial examining the effects of a two-year course of exenatide in people with Parkinson’s disease. Those data will be available in the second half of this year, according to Tom Foltynie, a neurologist at University College London, in comments provided to the UK Science Media Centre.
[ad_2]
Source link