When nucleic acids like DNA or RNA build up in a cell’s cytoplasm, it sets off an alarm call for the immune system. Enzymes usually clear these nucleic acids before they cause an issue, but when these enzymes don’t work and the immune system gets called in, it can lead to autoimmune and inflammatory diseases.
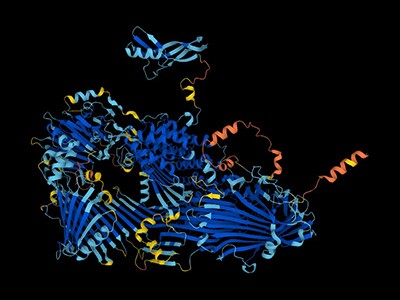
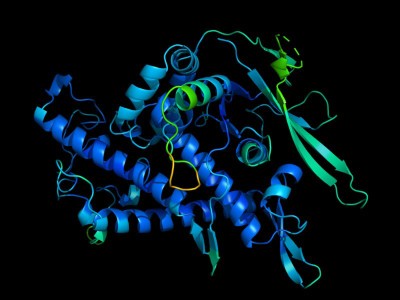
In a new study published on March 26, 2024 in the journal Structure, Scripps Research scientists present the previously undescribed structure of two of these nucleic acid-degrading enzymes-;PLD3 and PLD4. Understanding these enzymes’ structures and molecular details is an important step toward designing therapies for the various diseases that arise when they malfunction, which include lupus erythematosus, rheumatoid arthritis and Alzheimer’s disease.
These enzymes are important for cleaning up the cellular environment, and they also set the threshold for what is considered an infection or not. I’m hoping someday we may be able to help patients based on this information.”
David Nemazee, PhD, senior author, professor in the Department of Immunology and Microbiology at Scripps Research
Enzymes are proteins that speed up chemical reactions by binding and reacting to specific molecules called substrates. In the case of PLD3 and PLD4, the substrate is a strand of RNA or DNA, which the enzymes break down nucleotide by nucleotide.
The team used X-ray crystallography to build atomic-scale models of the PLD3 and PLD4 in multiple states or situations, allowing them to examine how their shapes changed over the course of the catalytic reaction. This included when the enzymes were resting, or when they were actively bound to a substrate.
“These models allow us to visualize PLD3 and PLD4 very clearly and with high resolution, so we know exactly how every atom interacts, meaning we can deduce how the enzymes work,” says first author Meng Yuan, a staff scientist in the Department of Integrative Structural and Computational Biology at Scripps Research.
The structural analyses revealed that PLD3 and PLD4 are structurally similar and that they degrade DNA and RNA in a very similar fashion, even though PLD4 is a larger protein. Both enzymes degrade nucleic acids via a two-step process.
“We call this process a two-step catalysis: bite down and release,” says Yuan. “In the first step, the enzyme bites down on the DNA strand and separates a single ‘brick’ or nucleotide from the rest of the strand, and in the second step, it opens its ‘mouth’ and releases the brick to be recycled.”
Because the enzymatic reaction happens so quickly-;within milliseconds-;researchers needed to use an alternative substrate to visualize the enzymes’ structure during catalysis. To do this, they incubated the enzymes together with a molecule that looks very similar to the DNA that the enzyme usually degrades, but that the enzymes degrade much more slowly.
This method uncovered a previously unknown function for one of the enzymes: In addition to biting off nucleotides from single-stranded RNA and DNA, PLD4 also showed phosphatase activity, which means it might also be involved in breaking down DNA’s phosphate backbone.
“I think it’s amazing that the crystal structure told us about this phosphatase activity,” says Nemazee. “To discover new enzymatic activity is unheard of in structural biology. It’s only because Meng was able to solve such an amazingly accurate and detailed structure that he could inform us about this extra enzymatic activity that we had no idea about.”
After they had elucidated PLD3 and PLD4’s usual structure, the researchers examined the structure of variants that are associated with diseases, including Alzheimer’s and spinocerebellar ataxia. These analyses revealed that some of these variants had decreased enzymatic capability, while others-;including a mutation associated with late-onset Alzheimer’s-;appeared to be more active.
“Some of our data suggests that one of these Alzheimer’s-associated enzyme variants might function better, which was a surprise to me, but it also may be less stable and more easily aggregated,” says Nemazee.
The researchers plan to continue investigating the structure and function of these enzymes. Their next steps include exploring possible ways of inhibiting the enzymes in scenarios where they are overactive, and they also plan to investigate the possibility of replacing the enzymes in people who carry non-functional (or non-working) versions.
In addition to Nemazee and Yuan, authors of the study, “Structural and mechanistic insights into disease-associated endolysosomal exonucleases PLD3 and PLD4,” were Linghang Peng, Deli Huang, Amanda Gavin, Fangkun Luan, Jenny Tran, Ziqi Feng, Xueyong Zhu, Jeanne Matteson, and Ian Wilson, all of Scripps Research.
This study was supported by the National Institutes of Health (grants R01AI142945 and RF1AG070775) and Skaggs Institute for Chemical Biology at Scripps Research.
Yuan, M., et al. (2024). Structural and mechanistic insights into disease-associated endolysosomal exonucleases PLD3 and PLD4. Structure. doi.org/10.1016/j.str.2024.02.019.
West, S., Gromak, N. & Proudfoot, N. J. Human 5′ -> 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature432, 522–525 (2004).
Fong, N. et al. Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol. Cell60, 256–267 (2015).
Krzyszton, M. et al. Defective XRN3-mediated transcription termination in Arabidopsis affects the expression of protein-coding genes. Plant J.93, 1017–1031 (2018).
Girbig, M., Misiaszek, A. D. & Muller, C. W. Structural insights into nuclear transcription by eukaryotic DNA-dependent RNA polymerases. Nat. Rev. Mol. Cell Biol.23, 603–622 (2022).
Porrua, O. & Libri, D. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Biol.16, 190–202 (2015).
Kumar, A., Clerici, M., Muckenfuss, L. M., Passmore, L. A. & Jinek, M. Mechanistic insights into mRNA 3′-end processing. Curr. Opin. Struc. Biol.59, 143–150 (2019).
Lunde, B. M. et al. Cooperative interaction of transcription termination factors with the RNA polymerase II C-terminal domain. Nat. Struct. Mol. Biol.17, 1195–1201 (2010).
Carminati, M. et al. A direct interaction between CPF and RNA Pol II links RNA 3′ end processing to transcription. Mol. Cell83, 4461–4478 (2023).
Brannan, K. et al. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol. Cell46, 311–324 (2012).
Jimeno-Gonzalez, S., Haaning, L. L., Malagon, F. & Jensen, T. H. The yeast 5′-3′ exonuclease Rat1p functions during transcription elongation by RNA polymerase II. Mol. Cell37, 580–587 (2010).
Cortazar, M. A. et al. Xrn2 substrate mapping identifies torpedo loading sites and extensive premature termination of RNA pol II transcription. Genes Dev.36, 1062–1078 (2022).
Kim, M., Ahn, S. H., Krogan, N. J., Greenblatt, J. F. & Buratowski, S. Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J.23, 354–364 (2004).
Zhang, Z., Fu, J. & Gilmour, D. S. CTD-dependent dismantling of the RNA polymerase II elongation complex by the pre-mRNA 3′-end processing factor, Pcf11. Genes Dev.19, 1572–1580 (2005).
Zhang, H., Rigo, F. & Martinson, H. G. Poly(A) signal-dependent transcription termination occurs through a conformational change mechanism that does not require cleavage at the poly(A) site. Mol. Cell59, 437–448 (2015).
Logan, J., Falck-Pedersen, E., Darnell, J. E. Jr & Shenk, T. A poly(A) addition site and a downstream termination region are required for efficient cessation of transcription by RNA polymerase II in the mouse beta maj-globin gene. Proc. Natl Acad. Sci. USA84, 8306–8310 (1987).
Eaton, J. D., Francis, L., Davidson, L. & West, S. A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev.34, 132–145 (2020).
Luo, W., Johnson, A. W. & Bentley, D. L. The role of Rat1 in coupling mRNA 3′-end processing to transcription termination: implications for a unified allosteric–torpedo model. Genes Dev.20, 954–965 (2006).
Cortazar, M. A. et al. Control of RNA Pol II speed by PNUTS-PP1 and Spt5 dephosphorylation facilitates termination by a “sitting duck torpedo” mechanism. Mol. Cell76, 896–908 e894 (2019).
Xue, Y. et al. Saccharomyces cerevisiaeRAI1 (YGL246c) is homologous to human DOM3Z and encodes a protein that binds the nuclear exoribonuclease Rat1p. Mol. Cell. Biol.20, 4006–4015 (2000).
Stevens, A. & Poole, T. L. 5′-exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5′-exonuclease-1. J. Biol. Chem.270, 16063–16069 (1995).
Grosso, A. R., de Almeida, S. F., Braga, J. & Carmo-Fonseca, M. Dynamic transitions in RNA polymerase II density profiles during transcription termination. Genome Res.22, 1447–1456 (2012).
Orozco, I. J., Kim, S. J. & Martinson, H. G. The poly(A) signal, without the assistance of any downstream element, directs RNA polymerase II to pause in vivo and then to release stochastically from the template. J. Biol. Chem.277, 42899–42911 (2002).
Gromak, N., West, S. & Proudfoot, N. J. Pause sites promote transcriptional termination of mammalian RNA polymerase II. Mol. Cell. Biol.26, 3986–3996 (2006).
James, K., Gamba, P., Cockell, S. J. & Zenkin, N. Misincorporation by RNA polymerase is a major source of transcription pausing in vivo. Nucleic Acids Res.45, 1105–1113 (2017).
Molodtsov, V., Wang, C., Firlar, E., Kaelber, J. T. & Ebright, R. H. Structural basis of Rho-dependent transcription termination. Nature14, 367–374 (2023).
Murayama, Y. et al. Structural basis of the transcription termination factor Rho engagement with transcribing RNA polymerase from Thermus thermophilus. Sci. Adv.9, eade7093 (2023).
Higo, T. et al. Development of a hexahistidine-3x FLAG-tandem affinity purification method for endogenous protein complexes in Pichia pastoris. J. Struct. Funct. Genomics15, 191–199 (2014).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods14, 290–296 (2017).
Farnung, L., Ochmann, M., Engeholm, M. & Cramer, P. Structural basis of nucleosome transcription mediated by Chd1 and FACT. Nat. Struct. Mol. Biol.28, 382–387 (2021).
Ehara, H., Kujirai, T., Shirouzu, M., Kurumizaka, H. & Sekine, S. I. Structural basis of nucleosome disassembly and reassembly by RNAPII elongation complex with FACT. Science377, eabp9466 (2022).
How memories are stored is an ongoing question in neuroscience. Now researchers have found an inflammatory pathway that responds to DNA damage in neurons has a key role in the persistence of memories. How this pathway helps memories persist is unclear, but the researchers suggest that how the DNA damage is repaired might play a role. As inflammation in the brain is often associated with disease, the team were surprised by this finding, which they hope will help uncover ways to better preserve our memories, especially in the face of neurodegenerative disorders.
Research Article: Jovasevic et al.
News and Views: Innate immunity in neurons makes memories persist
08:40 Research Highlights
The effect of wind turbines on property values, and how waste wood can be used to 3D print new wooden objects.
Research Highlight: A view of wind turbines drives down home values — but only briefly
Research Highlight: Squeeze, freeze, bake: how to make 3D-printed wood that mimics the real thing
11:14 How melting ice is affecting global timekeeping
Due to variations in the speed of Earth’s rotation, the length of a day is rarely exactly 24 hours. By calculating the strength of the different factors affecting this, a researcher has shown that although Earth’s rotation is overall speeding up, this effect is being tempered by the melting of the polar ice caps. As global time kept by atomic clocks occasionally has to be altered to match Earth’s rotation, human-induced climate change may delay plans to add a negative leap-second to ensure the two align.
Research article: Agnew
News and Views: Melting ice solves leap-second problem — for now
20:04 Briefing Chat
An AI for antibody development, and the plans for the upcoming Simons observatory.
Nature News: ‘A landmark moment’: scientists use AI to design antibodies from scratch
Nature News: ‘Best view ever’: observatory will map Big Bang’s afterglow in new detail
Subscribe to Nature Briefing, an unmissable daily round-up of science news, opinion and analysis free in your inbox every weekday.
Since the 1950s, scientists have had a pretty good idea of how muscles work. The protein at the centre of the action is myosin, a molecular motor that ratchets itself along rope-like strands of actin proteins — grasping, pulling, releasing and grasping again — to make muscle cells contract.
The basics were first explained in a pair of landmark papers in Nature1,2, and they have been confirmed and elaborated on by detailed molecular maps of myosin and its partners. Researchers think that myosin generates force by cocking back the long lever-like arm that is attached to the motor portion of the protein.
The only hitch is that scientists had never seen this fleeting pre-stroke state — until now.
In a preprint published in January3, researchers used a cutting-edge structural biology technique to record this moment, which lasts just milliseconds in living cells.
‘The entire protein universe’: AI predicts shape of nearly every known protein
“It’s one of the things in the textbook you sort of gloss over,” says Stephen Muench, a structural biologist at the University of Leeds, UK, who co-led the study. “These are experiments that people wanted to do 40 years ago, but they just never had the technology.”
That technology — called time-resolved cryo-electron microscopy (cryo-EM) — now has structural biologists thinking like cinematographers, turning still snapshots of life’s molecular machinery into motion pictures that reveal how it works.
Muench and his colleagues’ myosin movie isn’t feature-length; it consists of just two frames showing different stages of the molecular motion. Yet it confirmed a decades-old theory and settled debates over the order of the steps in myosin’s choreography. Other researchers are focusing their new-found director’s eye on understanding cell-signalling systems, including those underlying opioid overdoses, the gene-editing juggernaut CRISPR–Cas9 and other molecular machines that have been mostly studied with highly detailed, yet static structural maps.
Researchers have been able to capture images of individual myosin proteins as they pull on an actin filament during muscle contraction, confirming key details of the motion. First, myosin becomes cocked or primed, then it attaches to actin and its lever arm swings in a power stroke that slides the filament by about 34 nanometres.Credit: Sean McMillan
“The big picture is to move away, as much as possible, from this single, static snapshot,” says Georgios Skiniotis, a structural biologist at Stanford University in California, whose team used the technique to record the activation of a type of cell-signalling molecule called a G-protein-coupled receptor (GPCR)4. “I want the movie.”
Freeze frame
To underscore the power of cryo-EM, Skiniotis and others like to draw a comparison with one of the first motion pictures ever made. In the 1870s, photographer Eadweard Muybridge used high-speed photography technology, which was cutting edge at the time, to capture a series of still images of a galloping horse. They showed, for the first time, that all four of the animal’s hooves leave the ground at once — something that the human eye could not distinguish.
Similar insights, Skiniotis says, will come from applying the same idea to protein structures. “I want to get a dynamic picture.”
The ability to map proteins and other biomolecules down to the location of individual atoms has transformed biology, underpinning advances in gene editing, drug discovery and revolutionary artificial-intelligence tools such as AlphaFold, which can predict protein structures. But the mostly static images delivered by X-ray crystallography and cryo-EM, the two technologies responsible for the lion’s share of determined protein structures, belie the dynamic nature of life’s molecules.
“Biomolecules are not made up of rocks,” says Sonya Hanson, a computational biophysicist at the Flatiron Institute in New York City. They exist in water and are constantly in motion. “They’re more like jelly,” adds Muench.
The secret lives of cells — as never seen before
Biologists often say that “structure determines function”, but that’s not quite right, says Ulrich Lorenz, a molecular physicist at the Swiss Federal Institute of Technology in Lausanne (EPFL). The protein poses captured by most structural studies are energetically stable ‘equilibrium’ states that provide limited clues to the short-lived, unstable confirmations that are key to chemical reactions and other functions performed by molecular machines. “Structure allows you to infer function, but only incompletely and imperfectly, and you’re missing all of the details,” says Lorenz.
Cryo-EM is a great way to get at the details, but capturing these fleeting states requires careful preparation. Protein samples are pipetted onto a grid and then flash frozen with liquid ethane. They are then imaged using powerful electron beams that record snapshots of individual molecules (sophisticated software classifies and morphs these pictures into structural maps). The samples swim in water before being frozen, so any chemical reaction that can happen in a test tube can, in theory, be frozen in place on a cryo-EM grid — if researchers can catch it quickly enough.
That’s one of the first big challenges says Joachim Frank, a structural biologist at Columbia University in New York City who shared the 2017 Nobel Prize in Chemistry for his work on cryo-EM. “Even for very dexterous people, it takes a few seconds.” In that time, any chemical reactions — and the intermediate structures that mediate the reactions — might be long gone before freezing. “This is the gap we want to fill,” says Frank.
Caught in translation
Frank’s team has attempted to solve this problem using a microfluidic chip. The device quickly mixes two protein solutions, allows them to react for a specified time period and then delivers reaction droplets onto a cryo-EM grid that is instantly frozen.
This year, Frank’s team used their device to study a bacterial enzyme that rescues ribosomes, the cell’s protein-making factories, if they stall in response to antibiotics or other stresses. The enzyme, called HflX, helps to recycle stuck ribosomes by popping their two subunits apart.
Frank’s team captured three images of HflX bound to the ribosome, over a span of 140 milliseconds, which show how it splits the ribosome like someone carefully removing the shell from an oyster. The enzyme breaks a dozen or so molecular bridges that hold a ribosome’s two subunits together, one by one, until just two are left and the ribosome pops open5. “The most surprising thing to me is that it’s a very orderly process,” Frank says. “You would think the ribosome is being split and that’s it.”
Muench and his colleagues, including Charlie Scarff, a structural biologist at the University of Leeds, and Howard White, a kineticist at Eastern Virginia Medical School in Norfolk, Virginia also used a microfluidic chip to make their myosin movie by quickly mixing myosin and actin3.
‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures
But the molecular motor is so fast that, to slow things down ever further, they used a mutated version of myosin that operates about ten times slower than normal. This allowed the team to determine two structures, 110 milliseconds apart, that showed the swing of myosin’s lever-like arm. The structures also showed that a by-product of the chemical reaction that powers the motor — the breakdown of a cellular fuel called ATP — exits the protein’s active site before the lever swings and not after. “That is ending decades of conjecture,” says Scarff.
With this new model in mind, Scarff, whose specialty is myosin, and Muench are planning to use time-resolved cryo-EM to study how myosin dynamics are affected by certain drugs and mutations that are known to cause heart disease.
Microfluidic chips aren’t the only way researchers are putting time stamps on protein structures. A team led by Bridget Carragher, a structural biologist and the technical director at the Chan Zuckerberg Imaging Institute in Redwood City, California, developed a ‘spray and mix’ approach that involves shooting tiny volumes of reacting samples onto a grid before flash-freezing them6.
In another set-up — developed by structural physiologist Edward Twomey at Johns Hopkins University School of Medicine in Baltimore, Maryland, and his team — a flash of light triggers light-sensitive chemical reactions, which are stopped by flash-freezing7. Lorenz’s kit, meanwhile, takes already frozen samples and uses laser pulses to reanimate them for a few microseconds before they refreeze, all under the gaze of an electron microscope8.
‘Limitations everywhere’
The different approaches have their pros and cons. Carragher’s spray and mix approach uses minute sample volumes, which should be easy to obtain for most proteins; Twomey says his ‘open-source’ light-triggered device is relatively inexpensive and can be built for a few thousand dollars; and Lorenz says his laser-pulse system has the potential to record many more fleeting events than other time-resolved cryo-EM technologies — down to a tenth of a microsecond.
Revolutionary cryo-EM is taking over structural biology
But these techniques are not yet ready to be rolled out. Currently, there are no commercial suppliers of time-resolved cryo-EM technology, limiting its reach, says Rouslan Efremov, a structural biologist at the VIB-VUB Center for Structural Biology in Brussels. “All these things are fussy and hard to control and they haven’t really caught on,” adds Carragher.
Holger Stark, a structural biologist at the Max Planck Institute for Multidisciplinary Sciences in Göttingen, Germany, says that current forms of time-resolved cryo-EM might be useful for some molecular machines that operate on the basis of large-scale movements — for example, the ribosome. However, the technology is not ready for use on just any biological system. “You have to cherry pick your subject,” he says. “We have limitations everywhere.”
Despite the shortcomings, there are plenty of interesting questions for researchers to start addressing now using these techniques. Twomey is using time-resolved cryo-EM to study Cas9, the DNA-cutting enzyme behind CRISPR gene editing, and says the insights could help to make more efficient gene-editing systems.
Lorenz used his laser-melting method to show how a plant virus swells up after it infects a cell to release its genetic material7 (see ‘Viral blow-up’). He is now studying other viral entry molecules such as HIV’s envelope protein. “We have these static structures, but we don’t know how the system makes it from one state to the other, and how the machinery works,” he says.
Source: Ref.8
Skiniotis’s team is investigating GPCRs, including one called the β-adrenergic receptor, which has been implicated in asthma. Their work4 shows how activating the receptor triggers it to shed its partner G-protein, a key step in propagating signals in cells.
The researchers are now studying the same process in a GPCR called the µ-opioid receptor, which is activated by morphine and fentanyl among other drugs. In preliminary unpublished results, they have found that the dynamics of the receptor help to explain why some drugs such as fentanyl are so potent in promoting G-protein activation, while others aren’t. Such insights, says Skiniotis, are glimpses of unseen biology that molecular movies promise to reveal. Just don’t forget the popcorn.
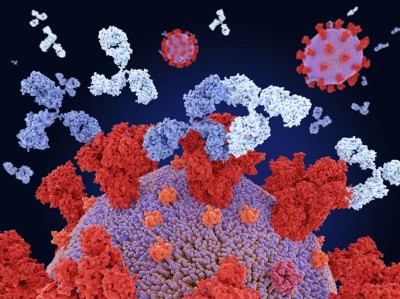
Antibodies (pink) bind to influenza virus proteins (yellow) (artist’s conception).Credit: Juan Gaertner/Science Photo Library
Researchers have used generative artificial intelligence (AI) to help them make completely new antibodies for the first time.
The proof-of-principle work, reported this week in a preprint on bioRxiv1, raises the possibility of bringing AI-guided protein design to the therapeutic antibody market, which is worth hundreds of billions of dollars.
AlphaFold touted as next big thing for drug discovery — but is it?
Antibodies — immune molecules that strongly attach to proteins implicated in disease — have conventionally been made using brute-force approaches that involve immunizing animals or screening vast numbers of molecules.
AI tools that can shortcut those costly efforts have the potential to “democratize the ability to design antibodies”, says study co-author Nathaniel Bennett, a computational biochemist at the University of Washington in Seattle. “Ten years from now, this is how we’re going to be designing antibodies.”
“It’s a really promising piece of research” that represents an important step in applying AI protein-design tools to making new antibodies, says Charlotte Deane, an immuno-informatician at the University of Oxford, UK.
Making mini proteins
Bennett and his colleagues used an AI tool that their team released last year2 that has helped to transform protein design. The tool, called RFdiffusion, allows researchers to design mini proteins that can strongly attach to another protein of choice. But these custom proteins bear no resemblance to antibodies, which recognize their targets by way of floppy loops that have proved difficult to model with AI.
To overcome this, a team co-led by computational biophysicist David Baker and computational biochemist Joseph Watson, both at the University of Washington, modified RFdiffusion. The tool is based on a neural network similar to those used by image-generating AIs such as Midjourney and DALL·E. The team fine-tuned the network by training it on thousands of experimentally determined structures of antibodies attached to their targets, as well as real-world examples of other antibody-like interactions.
How generative AI is building better antibodies
Using this approach, the researchers designed thousands of antibodies that recognize specific regions of several bacterial and viral proteins — including those that the SARS-CoV-2 and influenza viruses use to invade cells — and a cancer drug target. They then made a subset of their designs in the laboratory and tested whether the molecules could bind to the right targets.
Watson says that about one in 100 antibody designs worked as hoped — a lower success rate than the team now achieves with other types of AI-designed protein. The researchers determined the structure of one of the influenza antibodies, using a technique called cryo-electron microscopy, and found that it recognized the intended portion of the target protein.
Early proof of principle
A handful of companies are already using generative AI to help develop antibody drugs. Baker and Watson’s team hopes that RFdiffusion can help to tackle drug targets that have proved challenging, such as G-protein coupled receptors — membrane proteins that help to control a cell’s responses to external chemicals.
But the antibodies that RFdiffusion churned out are a long way from reaching the clinic. The designer antibodies that did work didn’t bind to their targets particularly strongly. Any antibody used therapeutically would also need its sequences modified to resemble natural human antibodies so as not to provoke an immune reaction.
The designs are also what’s known as single-domain antibodies, which resemble those found in camels and sharks, rather than the more complex proteins that nearly all approved antibody drugs are based on. These types of antibody are easier to design and simpler to study in the lab, and it makes sense to design these first, says Deane. “But this doesn’t take away from it being a step on the way to the kinds of methods we need.”
“This is proof-of-principle work,” Watson stresses. But he hopes this initial success will pave the way for designing antibody drugs at touch of a button. “It feels like quite a landmark moment. It really shows this is possible.”
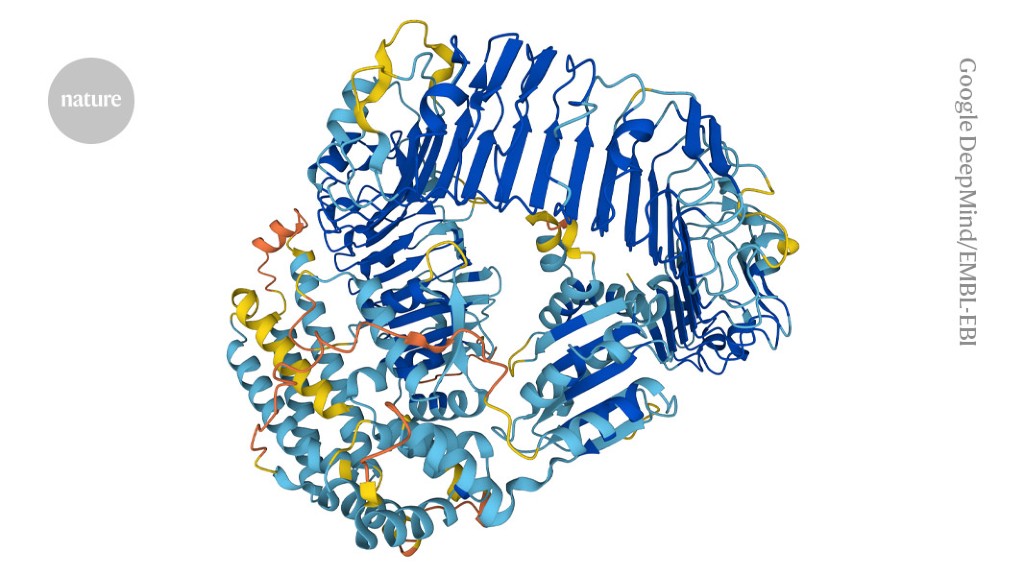
The artificial-intelligence tool AlphaFold can design proteins to perform specific functions.Credit: Google DeepMind/EMBL-EBI (CC-BY-4.0)
Could proteins designed by artificial intelligence (AI) ever be used as bioweapons? In the hope of heading off this possibility — as well as the prospect of burdensome government regulation — researchers today launched an initiative calling for the safe and ethical use of protein design.
“The potential benefits of protein design [AI] far exceed the dangers at this point,” says David Baker, a computational biophysicist at the University of Washington in Seattle, who is part of the voluntary initiative. Dozens of other scientists applying AI to biological design have signed the initiative’s list of commitments.
AI tools are designing entirely new proteins that could transform medicine
“It’s a good start. I’ll be signing it,” says Mark Dybul, a global health policy specialist at Georgetown University in Washington DC who led a 2023 report on AI and biosecurity for the think tank Helena in Los Angeles, California. But he also thinks that “we need government action and rules, and not just voluntary guidance”.
The initiative comes on the heels of reports from US Congress, think tanks and other organizations exploring the possibility that AI tools — ranging from protein-structure prediction networks such as AlphaFold to large language models such as the one that powers ChatGPT — could make it easier to develop biological weapons, including new toxins or highly transmissible viruses.
Designer-protein dangers
Researchers, including Baker and his colleagues, have been trying to design and make new proteins for decades. But their capacity to do so has exploded in recent years thanks to advances in AI. Endeavours that once took years or were impossible — such as designing a protein that binds to a specified molecule — can now be achieved in minutes. Most of the AI tools that scientists have developed to enable this are freely available.
To take stock of the potential for malevolent use of designer proteins, Baker’s Institute of Protein Design at the University of Washington hosted an AI safety summit in October 2023. “The question was: how, if in any way, should protein design be regulated and what, if any, are the dangers?” says Baker.
AlphaFold touted as next big thing for drug discovery — but is it?
The initiative that he and dozens of other scientists in the United States, Europe and Asia are rolling out today calls on the biodesign community to police itself. This includes regularly reviewing the capabilities of AI tools and monitoring research practices. Baker would like to see his field establish an expert committee to review software before it is made widely available and to recommend ‘guardrails’ if necessary.
The initiative also calls for improved screening of DNA synthesis, a key step in translating AI-designed proteins into actual molecules. Currently, many companies providing this service are signed up to an industry group, the International Gene Synthesis Consortium (IGSC), that requires them to screen orders to identify harmful molecules such as toxins or pathogens.
“The best way of defending against AI-generated threats is to have AI models that can detect those threats,” says James Diggans, head of biosecurity at Twist Bioscience, a DNA-synthesis company in South San Francisco, California, and chair of the IGSC.
Risk assessment
Governments are also grappling with the biosecurity risks posed by AI. In October 2023, US President Joe Biden signed an executive order calling for an assessment of such risks and raising the possibility of requiring DNA-synthesis screening for federally funded research.
Baker hopes that government regulation isn’t in the field’s future — he says it could limit the development of drugs, vaccines and materials that AI-designed proteins might yield. Diggans adds that it’s unclear how protein-design tools could be regulated, because of the rapid pace of development. “It’s hard to imagine regulation that would be appropriate one week and still be appropriate the next.”
But David Relman, a microbiologist at Stanford University in California, says that scientist-led efforts are not sufficient to ensure the safe use of AI. “Natural scientists alone cannot represent the interests of the larger public.”
Vassallo, C. N., Doering, C. R., Littlehale, M. L., Teodoro, G. I. C. & Laub, M. T. A functional selection reveals previously undetected anti-phage defence systems in the E. coli pangenome. Nat. Microbiol.7, 1568–1579 (2022).
Kazlauskiene, M., Kostiuk, G., Venclovas, Č., Tamulaitis, G. & Siksnys, V. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science357, 605–609 (2017).
Lowey, B. et al. CBASS immunity uses CARF-related effectors to sense 3′–5′- and 2′–5′-linked cyclic oligonucleotide signals and protect bacteria from phage infection. Cell182, 38–49 (2020).
Ka, D., Oh, H., Park, E., Kim, J.-H. & Bae, E. Structural and functional evidence of bacterial antiphage protection by Thoeris defense system via NAD+ degradation. Nat. Commun.11, 2816 (2020).
Fernández-Lucas, J. et al. Biochemical and structural studies of two tetrameric nucleoside 2′-deoxyribosyltransferases from psychrophilic and mesophilic bacteria: Insights into cold-adaptation. Int. J. Biol. Macromol.192, 138–150 (2021).
Armstrong, S. R., Cook, W. J., Short, S. A. & Ealick, S. E. Crystal structures of nucleoside 2-deoxyribosyltransferase in native and ligand-bound forms reveal architecture of the active site. Structure4, 97–107 (1996).
Zhang, Q., Bhattacharya, S. & Andersen, M. E. Ultrasensitive response motifs: basic amplifiers in molecular signalling networks. Open Biol.3, 130031 (2013).
Park, C. K. & Horton, N. C. Structures, functions, and mechanisms of filament forming enzymes: a renaissance of enzyme filamentation. Biophys. Rev.11, 927–994 (2019).
Zaremba, M. et al. Short prokaryotic Argonautes provide defence against incoming mobile genetic elements through NAD+ depletion. Nat. Microbiol.7, 1857–1869 (2022).
Clabbers, M. T. B. et al. MyD88 TIR domain higher-order assembly interactions revealed by microcrystal electron diffraction and serial femtosecond crystallography. Nat. Commun.12, 2578 (2021).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D75, 861–877 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods14, 290–296 (2017).
• Ultrashort, intense X-ray pulses generated at facilities known as X-ray free-electron lasers (XFELs) have been used to probe light-induced structural changes in proteins.
• Light-responsive proteins typically absorb one optical or ultraviolet photon in natural settings, but could absorb more from the intense ‘pump’ lasers used to induce structural changes in these studies.
• Such unnatural absorption of multiple pump photons might force proteins to behave in ways that are not biologically relevant.
• Questions have therefore been raised about how these studies should be interpreted.
• Barends et al.1 now show that the structure of a model protein changes in different ways depending on whether single or multiple photons are absorbed.
RICHARD NEUTZE: Imperfect experiments can be informative
Structural changes that occur in proteins during biochemical reactions can be measured using a technique called time-resolved X-ray diffraction (TR-XRD). In this method, reactions are initiated in protein crystals, and X-ray pulses are used to record X-ray diffraction data at selected times after initiation. TR-XRD has produced structural insights into the pathways of diverse biological processes2, including photosynthesis, sensory signalling, ion transport and photodissociation — the light-induced breakage of bonds between proteins and their ligand molecules.
Read the paper: Influence of pump laser fluence on ultrafast myoglobin structural dynamics
For light-sensitive proteins, a pump laser pulse is used to initiate the reaction of interest. All molecules probed in a crystal contribute to the measured X-ray diffraction pattern, yet typically only a subpopulation is activated by the pump laser. A quantity known as the crystallographic occupancy estimates the fraction of molecules in a crystal that are activated. Raising the pump-laser fluence — the energy delivered per unit area by the pump laser onto a crystal — can increase the crystallographic occupancy, but more than one photon can be absorbed by the protein at high laser fluences3,4.
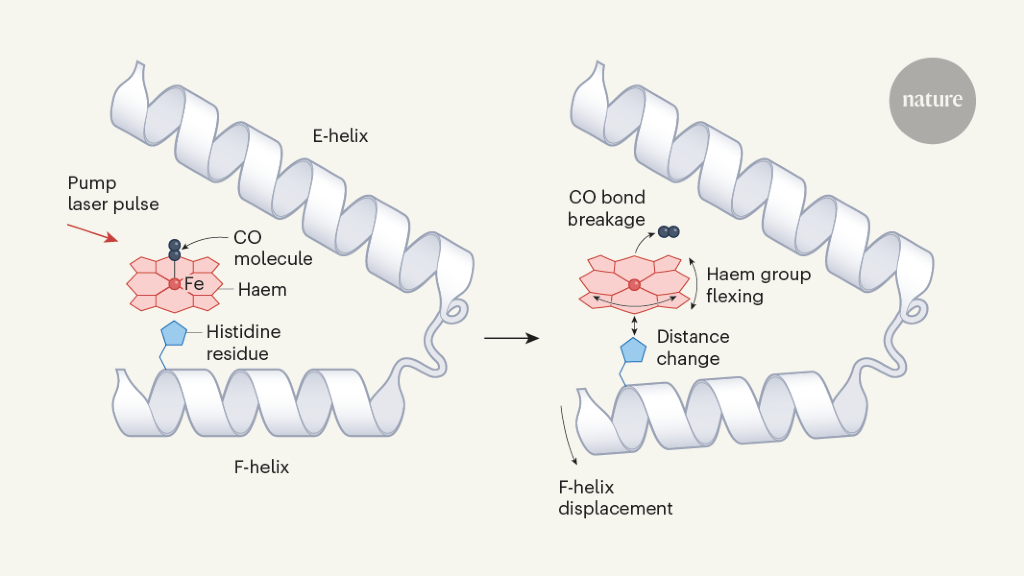
Barends et al. studied structural changes that occur in the carbon monoxide complex of the protein myoglobin (MbCO) after pump-laser-induced photodissociation of CO from the iron atom of a haem group (Fig. 1). This process was previously studied using TR-XRD at time resolutions of 7.5 nanoseconds5 and 150 picoseconds (1 ps is 10–12 seconds)6 using relatively large protein crystals (dimensions in the range of about 0.1 to 0.3 millimetres) and X-ray pulses from a synchrotron facility, which is a less intense X-ray source than an XFEL.
Figure 1 | Structural dynamics of a model protein system. Barends et al.1 used a method called time-resolved X-ray diffraction (TR-XRD) to study crystals of the carbon monoxide complex of the protein myoglobin (MbCO; only part of the complex is shown). They used a ‘pump’ laser pulse to induce photodissociation (breakage of the bond between CO and the iron (Fe) atom of a haem group in the protein), and then used ultrashort, intense X-ray pulses to obtain X-ray structures of the protein over time. Three structural changes crucial to photodissociation are shown (right): flexing of the haem group; changes in the distance between the haem and a histidine amino-acid residue in the F α-helix of the protein; and displacement of the F-helix. The authors observed that the fluence of the pump laser (the energy delivered per unit area by the pump laser onto a crystal) alters the amplitude and timing of these motions, and also the motion of the CO away from the haem after photodissociation — suggesting that lower fluences are needed to observe structural changes that occur in natural settings.
A 2015 study by some of the same researchers as Barends et al. used extremely short, intense XFEL pulses to record TR-XRD data from tens of thousands of much smaller MbCO crystals (average size 15 micrometres × 5 µm × 3 µm). This thereby achieved a time resolution of 250 femtoseconds (1 fs is 10–15 s) and revealed ultrafast conformational changes of the protein as photodissociation occurs7. But because those experiments used a high pump-laser fluence, Barends et al. have now repeated their study using a lower fluence that ensures single-photon excitation of MbCO.
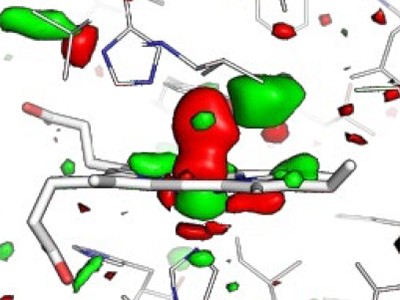
The authors used their TR-XRD data to determine difference Fourier maps, which show differences in electron density in MbCO before and after activation. Barends et al. found that lower pump-laser fluences yield lower crystallographic occupancies in maps produced 10 ps after protein activation. For this time delay, differences between structural changes induced by single-photon and multiphoton excitation are difficult to see from the difference Fourier maps, but the authors argue that some effects can be determined from careful analysis and structural modelling. A caveat is that structural modelling incurs larger errors when the crystallographic occupancy is low.
Clues to how water splits during photosynthesis
For sub-picosecond time delays, the authors’ analysis shows that the protein responds unexpectedly rapidly to the highest pump-laser fluence, and that CO swiftly populates a new location near the haem (Fig. 1). By contrast, it takes longer for the protein to respond and for this CO site to be populated after single-photon activation.
Barends and colleagues’ recording of high-quality maps from very small crystals of MbCO after single-photon excitation is impressive. Their study will motivate other researchers to recover high-quality difference Fourier maps using single-photon excitation. However, this might not be possible for many biological systems. Crucially, the structural perturbations of MbCO induced by single photons are in keeping with findings from earlier work5–7, illustrating that useful insight does emerge from imperfect experiments. Historically, the field has chosen not to let the perfect be the enemy of the good. I contend that such pragmatism should continue, so that the diversity of biological systems studied by TR-XRD continues to grow2.
R. J. DWAYNE MILLER: Protein ‘music’ must not be distorted
Biological processes are driven by chemistry. Chemistry is dynamic, with all the interconnecting atoms in molecules jiggling around, vying for right of way to take part in reaction pathways. In proteins, chemical transformations occur at an active or binding site. These processes involve the coupling of reaction forces such that some 10–100 atoms at the active site direct the motions of the 1,000–10,000 (or more) atoms of the surrounding protein. An astronomical number of possible conformational pathways (sequences of molecular structures) can occur, but only one or a few motions at the active site — known as reaction modes — direct biological function.
How can such a small number of reaction modes preferentially direct protein functions within the ocean of alternative conformational pathways? This apparent paradox is solved by considering spatially correlated motions — these arise when forces imprinted in the protein structure cause atoms to move together, rather than randomly and independently. By directly observing atomic motions during the defining moments of a chemical reaction, the initial distillation of all possible pathways down to a few reaction modes can be seen8. Such observations allow relationships between protein structure and function to be determined, but only if the reaction is initiated correctly. The whole point of such studies is that we don’t know the length and timescales of protein responses to the chemical driving force.
To help make sense of this issue, consider the fluid that forms by mixing cornflour and water. If you dip your finger slowly into this mixture, the fluid flows around it like a liquid. But if you rapidly poke the mixture, the material responds as an elastic solid. Similarly, the spatially varying barriers to motions in proteins mean that protein responses to forces depend on both the time and the length scales of the applied force. Given that protein structures are highly anisotropic (different in different directions), the response will also depend on the spatial location of the induced force.
Earliest molecular events of vision revealed
Until the work of Barends et al., femtosecond TR-XRD studies had used extremely high pump fluences to ensure that structural changes in proteins could be observed. Because the laser pulses are so short, this came at the cost of inducing multiphoton protein activation that produces initial atomic displacements different to those arising from single-photon activation, and at higher energies, with the atoms being displaced farther from their equilibrium positions4. The driving forces for the observed structural changes were therefore both spatially different and significantly larger than those of the biological pathway of interest4.
For the model MbCO system, there are three biologically crucial and spatially coupled motions (Fig. 1): the haem doming coordinate (which describes the flexing of the protein’s haem group); movement of a histidine amino-acid residue positioned close to the haem; and the displacement of α-helices resulting from the transfer of driving forces from the first two motions. Barends et al. observed that multiphoton activation resulted in much faster displacement of the histidine than did single-photon activation, and led to similarly faster motions of one of the helices. Moreover, the spatially correlated helical motions extended over different distances along the helix than did those induced by single-photon activation, and subsequently dissipated to different degrees.
The impulsive nature of the force produced by multiphoton activation on the initial histidine motion, and the temporal evolution of the positioning of the CO molecule in the protein pocket that contains the haem, are evidence of a different reaction pathway to the one produced by single-photon activation. There are significant differences in the magnitude, temporal evolution and pathway of the structural dynamics throughout the protein.
Given the importance of energetics and spatially correlated motions for understanding the structural changes that underpin protein function, we need to get this right. Biologically relevant protein motions are like music, albeit playing at frequencies we can’t hear. We need to observe the motions that correspond to certain frequencies, and discern how those motions are damped to produce net displacements. Imagine listening to music in which violin strings are struck too strongly, causing the accidental playing of different chords together. You would not hear the music as written, but something else entirely. To understand how nature works, we need to listen to the molecular music as nature wrote it. Barends et al. have done just that.
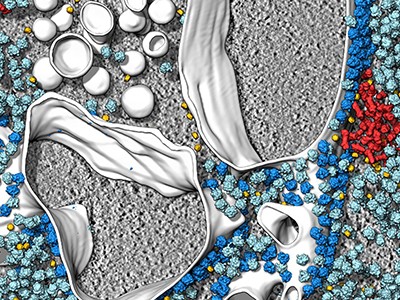
A recent re-emergence and outbreak of Mpox brought poxviruses back as a public health threat, underlining an important knowledge gap at their core. Now, a team of researchers from the Institute of Science and Technology Austria (ISTA) lifted the mysteries of poxviral core architecture by combining various cryo-electron microscopy techniques with molecular modeling. The findings, published in Nature Structural & Molecular Biology, could facilitate future research on therapeutics targeting the poxvirus core.
Variola virus, the most notorious poxvirus and one of the deadliest viruses to have afflicted humans, wreaked havoc by causing smallpox until it was eradicated in 1980. The eradication succeeded thanks to an extensive vaccination campaign using another poxvirus, the aptly named Vaccinia virus. The 2022-2023 re-emergence and outbreak of Mpox virus reminded us once more that viruses find ways to return to the forefront as public health threats. Importantly, this has highlighted the fundamental questions about poxviruses that have remained unanswered to this day.
One such fundamental question lies, quite literally, at the core of the matter: “We know that for poxviruses to be infective, their viral core must be properly formed. But what is this poxviral core made of, and how do its individual components come together and function?” asks ISTA Assistant Professor Florian Schur, the corresponding author of the study.
Schur and his team now put their finger on the missing link: a protein called A10. Interestingly, A10 is common to all clinically relevant poxviruses. In addition, the researchers found that A10 acts as one of the main building blocks of the poxviral core. This knowledge could be instrumental for future research on therapeutics targeting the poxviral core.
“The most advanced cryo-EM techniques available today”
The viral core is one of the factors common to all infectious poxvirus forms.
Previous experiments in virology, biochemistry, and genetics suggested several core protein candidates for poxviruses, but there were no experimentally-derived structures available.”
Julia Datler, ISTA PhD student, one of the co-first authors of the study
Thus, the team started by computationally predicting models of the main core protein candidates, using the now-famous AI-based molecular modeling tool AlphaFold. In parallel, Datler was setting the project’s biochemical and structural foundations by drawing on her background in virology and the Schur group’s main expertise: cryogenic electron microscopy, or cryo-EM for short. “We integrated many of the most advanced cryo-EM techniques available today with AlphaFold molecular modeling. This gave us, for the first time, a detailed overall view of the poxviral core–the ‘safe’ or ‘bioreactor’ inside the virus that encloses the viral genome and releases it in infected cells,” says Schur. “It was a bit of a gamble, but we eventually managed to find the right mix of techniques to examine this complex question,” says postdoc Jesse Hansen, the study’s co-first author whose expertise in various structural biology techniques and image processing methods was pivotal for the project.
A global 3D view of the poxvirus
The ISTA researchers examined “live” Vaccinia virus mature virions and purified poxviral cores under every possible angle–quite literally. “We combined the ‘classic’ single-particle cryo-EM, cryo-electron tomography, subtomogram averaging, and AlphaFold analysis to gain an overall view of the poxviral core,” says Datler. With cryo-electron tomography, researchers can reconstitute 3D volumes of a biological sample as large as an entire virus by acquiring images while gradually tilting the sample. “It’s like doing a CT scan of the virus,” says Hansen. “Cryo-electron tomography, our lab’s ‘specialty,’ allowed us to gain nanometer-level resolutions of the whole virus, its core, and interior,” says Schur. In addition, the researchers could fit the AlphaFold models into the observed shapes like a puzzle and identify molecules that make up the poxviral core. Among these, the core protein candidate A10 stood out as one of the major components. “We found that A10 defines key structural elements of the core of poxviruses,” says Datler. Schur adds, “These findings are a great resource to interpret bits of structural and virological data generated over the last decades.”
A rugged path to uncovering poxviral cores
The path to these findings was all but straightforward. “We needed to find our own way from the start,” says Datler. Leveraging her expertise in biochemistry, virology, and structural biology, Datler isolated, propagated, and purified samples of Vaccinia virus and established the protocols to purify the complete viral core, all while optimizing these samples for structural studies. “Structurally, it was extremely hard to study these virus cores. But luckily, our perseverance and optimism paid off,” says Hansen.
The ISTA researchers are convinced that their findings could provide a knowledge platform for future therapeutics that seek to target poxviral cores. “For example, one could think of drugs that prevent the core from assembling – or even disassembling and releasing the viral DNA during infection. Ultimately, fundamental virus research, as done here, allows us to be better prepared against possible future viral outbreaks,” concludes Schur.
Datler, J., et al. (2024). Multi-modal cryo-EM reveals trimers of protein A10 to form the palisade layer in poxvirus cores. Nature Structural & Molecular Biology. doi.org/10.1038/s41594-023-01201-6.