[ad_1]
Proteins are important molecules—but they aren’t the only molecules in a cell, and they don’t operate alone. In a new preprint, the team behind the protein structure prediction software RoseTTAFold has announced a tool that expands the types of chemistry that protein designers using deep learning will be able to incorporate, to reflect proteins’ environment better (bioRxiv 2023, DOI: 10.1101/2023.10.09.561603).
Protein structure prediction algorithms, such as AlphaFold and RoseTTAFold, have swept through the field of structural biology in recent years. These machine learning tools, trained on protein structures that have been solved experimentally, predict new 3D structures based only on proteins’ amino acid sequences. Biochemists use those predictions to develop hypotheses about how proteins work and how they fit together, and they have also used the tools to design new proteins with desired functions.
The trouble is, these models overlook many types of chemistry that can influence a protein’s structure. “A lot of biology involves, for example, proteins interacting with small molecules,” says senior author David Baker, a University of Washington professor whose lab developed RoseTTAFold. The latest update to the model, RoseTTAFold All-Atom, can handle the more diverse chemistry that occurs when proteins bind small molecules or undergo covalent modifications that can dramatically influence their structure and function.
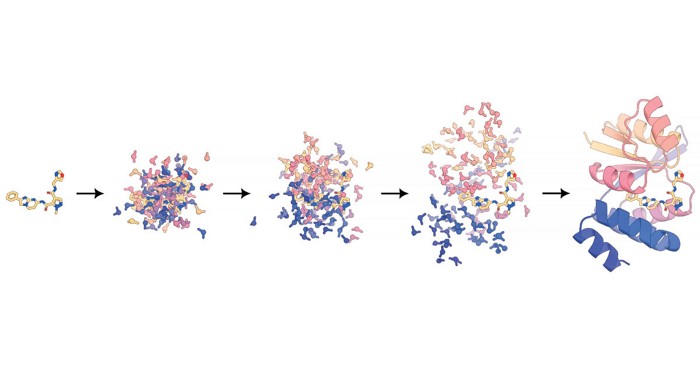
“It’s kind of a hypothesis that we wanted to test: Is it possible, even, to train a model that can represent all these different types of molecules?” says Rohith Krishna, a graduate student and first author of the paper. After about 2 years of tinkering, he and his colleagues found that they could, by combining two different modeling approaches for molecules. Like previous RoseTTAFold iterations, the new network represents polymer subunits, like amino acids or nucleobases, as single units, but it also represents every atom as a unit in each covalent modification and small-molecule binding partner. It’s hard to say exactly how it works—machine learning models are notoriously opaque—but the researchers think that it arranges all the units present until it reaches a plausible structure.
While prior updates to RoseTTAFold focused on specific problems, like predicting how proteins and nucleic acids interact, the team trained this one more broadly. “In principle, a network that’s been trained on more diverse sets of data should be able to generalize better,” Baker says, adding that the researchers plan for this network to supersede task-specific versions of RoseTTAFold.
The team used RoseTTAFold All-Atom to generate more-accurate predictions of proteins, such as enzymes bound to both their substrates and cofactors, and proteins with numerous covalent modifications. They also designed from scratch several proteins that bind to small molecules, using only the small molecules as inputs. Though he hasn’t tried more complex arrangements yet, Krishna predicts that the model may be able to help design proteins that undergo complicated interactions—for example with both a nucleic acid and a small molecule cofactor.
According to Lauren Porter, a computational biologist at the National Library of Medicine, the network is promising, but it will take time to see just how dramatic an advance it represents and where it might fall short. AI models in general, she says, are “only as good as their training set.” And they can stumble in areas where limited training data are available, such as when a single protein adopts two substantially different shapes depending on its context. Biochemists themselves aren’t aware of many of those cases, so it takes time for these discrepancies to be uncovered. Still, Porter says, “it’s a step in the right direction, for sure, and maybe a big one—time will tell.”
[ad_2]
Source link
Leave a Reply