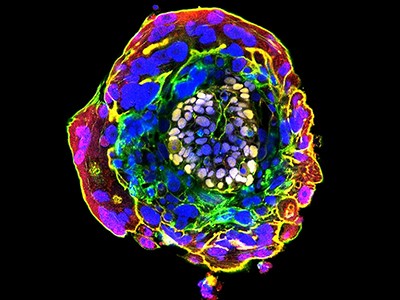
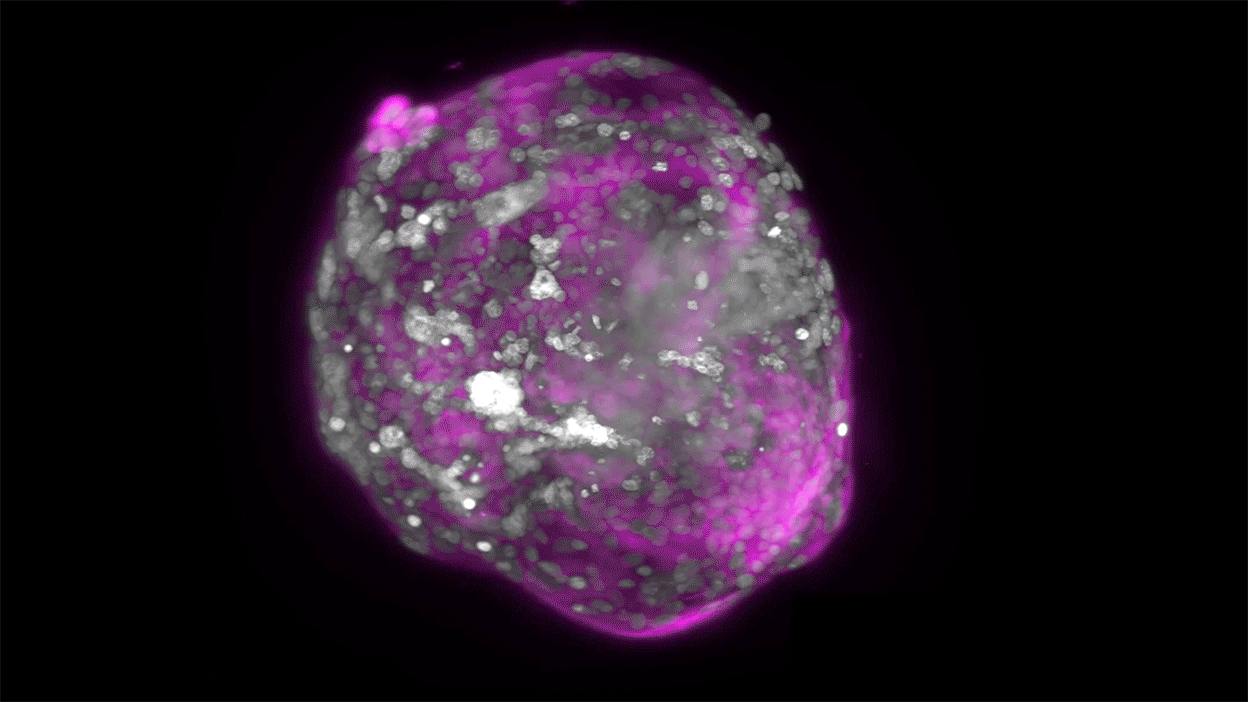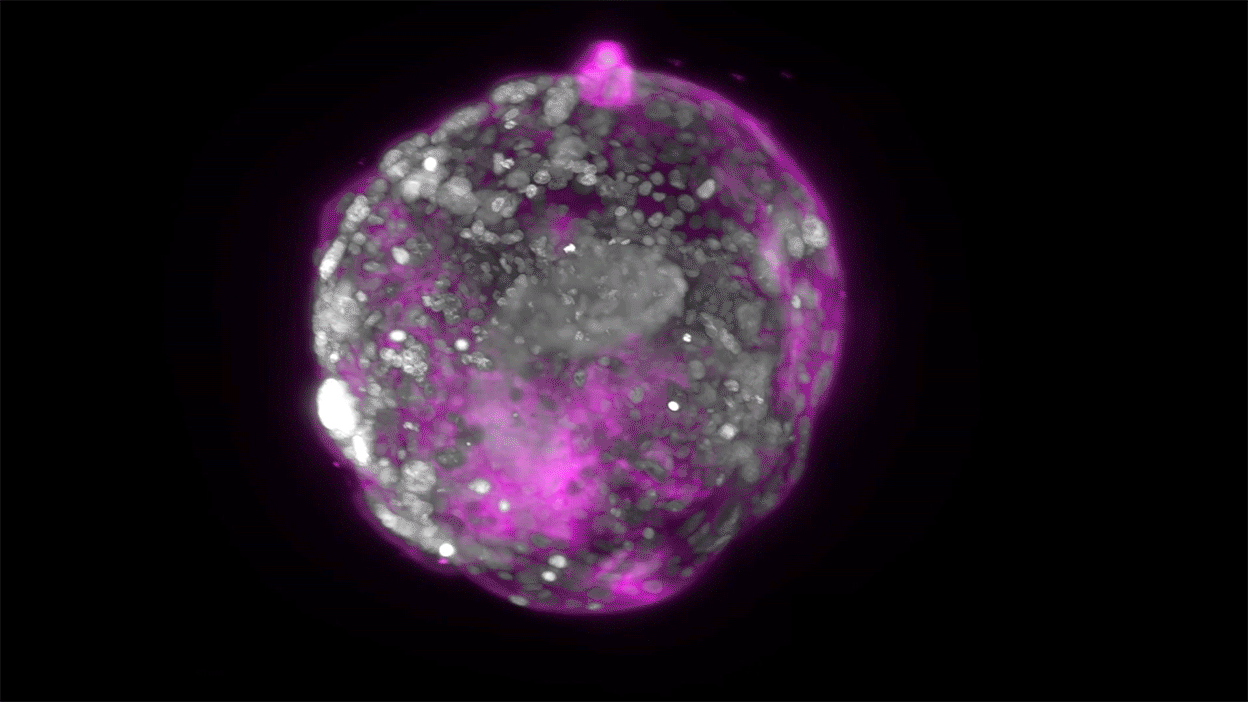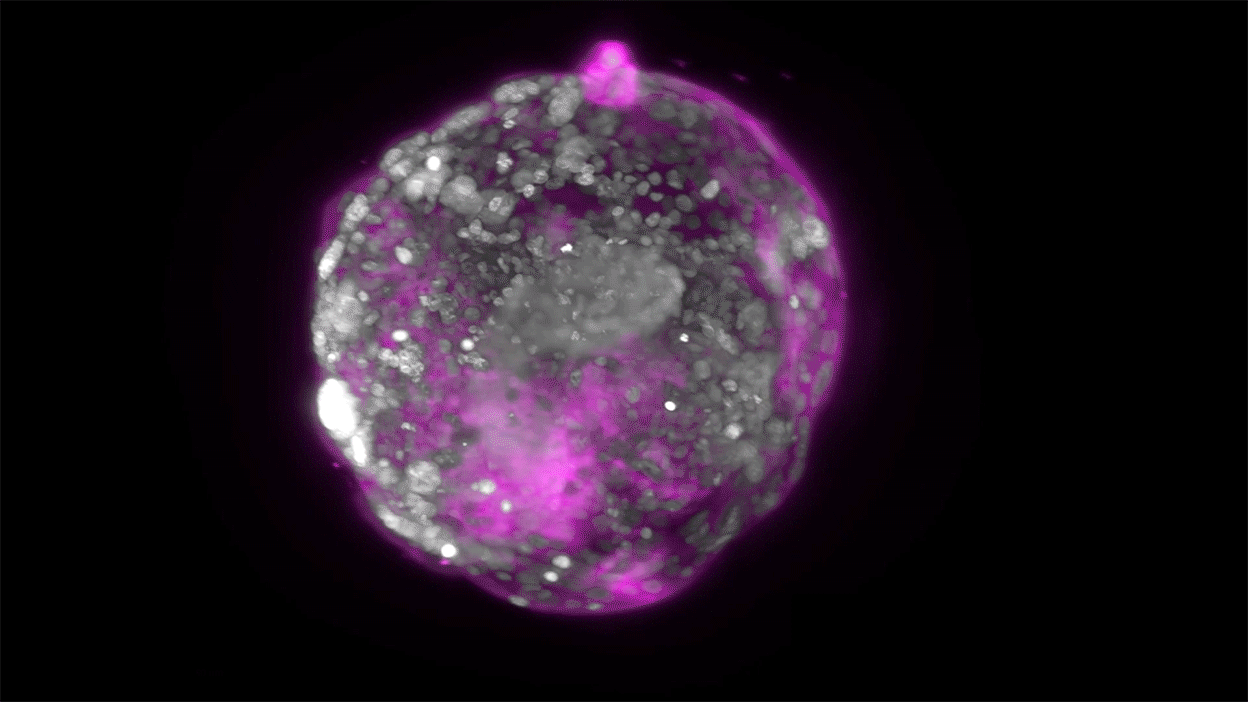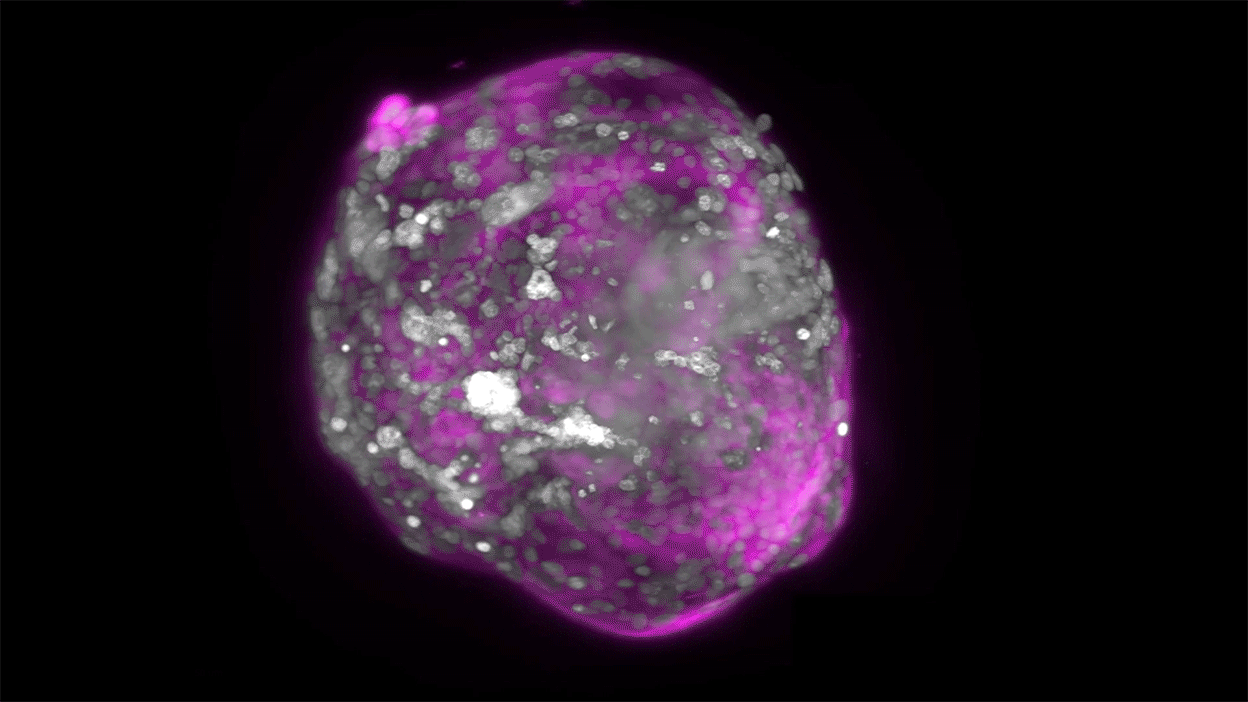
Embryo recovery and documentation
All of the animal procedures were approved by the Institutional Animal Care and Use Committee and were performed in strict adherence to Weizmann Institute guidelines. Mice were monitored for health and activity and were given ad libitum access to water and standard mouse chow with 12 h–12 h light–dark cycles. Embryos were collected from timed pregnant immune-competent C57BL/6JRccHsd or Hsd:ICR(CD-1) female mice (obtained from Envigo and mated in house with males of the same strain) between E5.5 and E9.5. Embryos were recovered from their implantation sites using fine forceps, in PBS, while carefully preserving all extraembryonic tissues. The embryos were then washed in PBS and transferred to chilled DMEM (Phenol-red free, GIBCO) supplemented with 10% FBS (Biological Industries) for imaging before dissociation. Phase-contrast images were taken using the Eclipse Ti2 inverted microscope (Nikon) and Zyla sCMOS camera (Andor). Morphological staging and analysis of embryos was conducted as previously described61.
Cell line information
Source of cell lines: all of the cell lines used in this study were generated in-house from a stock of validated V6.5 (C57BL/6×129) background. Authentication of cell lines: the cell lines were authenticated using genotyping PCR to ensure their identity and purity. The genotyping results confirmed the expected genetic background for each cell line. Mycoplasma contamination testing: all of the cell lines were routinely tested for mycoplasma contamination using the PCR-based Mycoplasma Detection Kit (Hylab) before use in the experiments. No mycoplasma contamination was detected in any of the cell lines.
Trophectoderm-specific genetic manipulation
Production of lentiviral vectors
Lentiviral vectors were constructed to produce lentiviruses expressing gRNAs designed to selectively target either GFP expression and the third exon of Bmp4 or the third and fourth exons of the Elf5 gene locus in the trophectoderm of Cas9–GFP62 mouse blastocysts. All gRNAs were selected for minimal off-target effects using the CCTop CRISPR/Cas9 target online predictor (https://cctop.cos.uni-heidelberg.de:8043/)63. Introduction of a mega-primer (Supplementary Table 4) that includes gRNAs into a lentivector, constitutively expressing gRNAs scaffolds and a mCherry fluorescent reporter, was carried out by restriction-free cloning as previously described64. Recombinant lentiviruses were produced by transient transfection into HEK293T cells, using polyethylene imine (PEI) (PEI linear, Mr 25,000, Polyscience) as previously described65, using three envelope and packaging plasmids and one of three viral constructs: (1) pDecko-GFP/mCherry (that is, the control vector), (2) pDecko-Elf5/mCherry or (3) pDecko-Bmp4/mCherry. In brief, infectious lentiviruses were collected at 48 and 72 h after transfection, filtered through 0.45-mm-pore cellulose acetate filters and concentrated by ultracentrifugation at 20,000 rpm for 2 h. Lentiviral supernatant effective titres were determined by infection of HEK293T cells followed by fluorescence-activated cell sorting (FACS) analysis. To validate of Elf5 and Bmp4 KO, HEK293T cells were infected with the appropriate lentiviral vector expressing gRNAs targeting Elf5/Bmp4 and mCherry. Infected cells were picked by sterile sorting, subsequently transfected with px330 Cas9 targeting plasmid expressing GFP66 and sorted again prior to DNA extraction. Genomic DNA was extracted by PCR-compatible lysis buffer (10 mM Tris, pH 8, 0.45% Triton X-100, 0.45% Tween-20, 0.2 mg ml−1 proteinase K). Primers flanking the PAM sequence of each target (Supplementary Table 4) were used for amplifying the genomic segments that included the expected Cas9-mediated DNA editing, and immediately followed by Sanger sequencing (not shown).
Mice and lentiviral transduction
Infection of nascent blastocysts was performed using B6D2F1 (C57BL/6xDBA) (Envigo)/Cas9-GFP embryos. In brief, 3–4-week old B6D2F1 female mice were hormone primed by an intraperitoneal injection of pregnant mare serum gonadotropin (PMSG, Vetmarket) followed 46 h later by an injection of human chorionic gonadotropin (hCG, Sigma-Aldrich) and mating with homozygote Cas9-GFP males62. Embryos were collected at the zygote stage, and cultured in KSOM medium until the blastocyst stage. For efficient infection of the trophectoderm, zona pellucida was removed in acidic Tyrode’s solution (Sigma-Aldrich)27. Next, 15–20 embryos were incubated with lentiviruses, described above, in KSOM for 4–5 h. The transduced blastocysts were washed, and then were transferred into each recipient female generated after mating with vasectomized CD1 males (Envigo); the day of injection was considered to be 2.5 days post coitum. Mice were handled in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee (IACUC, The Weizmann Institute of Science).
Analysis of genetically manipulated embryos
Elf5-KO embryos were collected at E7.5, and Bmp4-KO embryos were collected in a time series from E7.5 to 8.5, and all were dissected in ice-cold 1× PBS. Individual mutants were imaged using the Eclipse Ti2 inverted microscope (Nikon) and the Zyla sCMOS camera (Andor) while being maintained in DMEM supplemented with 10% FBS. Embryos positive for mCherry were selected for further morphological and transcriptome analysis (only in Elf5-KO embryos).
Tetraploid complementation assay for generating embryonic Bmp4 KO
Bmp4LoxP/LoxP embryonic stem cells were derived from Bmp4LoxP/LoxP mice67 (C57BL/6×129; Extended Data Fig. 11a) using standard embryonic stem cell derivation method. The cells were then treated with recombinant His-TAT-NLS-Cre (HTNC) protein (Addgene plasmid, 13763). For genotyping, individual clones were grown for two passages on gelatin-coated plates to eliminate residual MEF, and RNA was extracted using Direct-zol (Zymo RNA miniPrep, R2052) followed by cDNA production and quantitative PCR (qPCR; Supplementary Table 4). For this study, we used two validated Bmp4Δ/Δ clones and one isogenic control clone (a Bmp4+/+ HTNC-treated clone). Blastocyst injections were performed using (C57BL/6xDBA) B6D2F1 (Envigo) host embryos. In brief, 3–4-week old B6D2F1 females were hormone primed by an intraperitoneal injection of pregnant mare serum gonadotropin (PMSG, Vetmarket) followed 46 h later by an injection of human chorionic gonadotropin (hCG, Sigma-Aldrich). Embryos were collected at the zygote stage, and cultured in a CO2 incubator until the blastocyst stage. For tetraploid complementation, two-cell embryos were fused to one cell using a CF150/F instrument (BLS), by 2 DC square pulses of 30 V 40 ms and 1–2 V AC, in 0.3 M mannitol solution with BSA. On the day of the injection, embryos were placed in M2 medium using a 16-µm-diameter injection pipet (Biomedical Instruments) and a Piezo micromanipulator (Prime Tech); approximately 15 cells were injected into the blastocoel of each embryo. Approximately 20 blastocysts were transferred to each recipient female (CD1 female mice, Envigo); the day of injection was considered to be E2.5. Mice were handled in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee (IACUC, The Weizmann Institute of Science).
Ex utero culture of post-implantation embryos
Pregastrulating embryos were dissected at E5.5, their Reichert’s membrane removed and individually placed into separate wells of 8-well glass-bottom ibiTreat μ-plates (iBidi; 80827/80826) filled with 250 μl of EUCM (consisting of 25% DMEM (GIBCO 11880; includes 1 mg ml−1 d-glucose and pyruvate, without phenol red and without l-glutamine) supplemented with 1× GlutaMax (GIBCO, 35050061), 100 U ml−1 penicillin–100 μg ml−1 streptomycin (Biological industries; 030311B) and 11 mM HEPES (GIBCO, 15630056), plus 50% rat serum (rat whole-embryo culture serum, ENVIGO Bioproducts B-4520) and 25% human umbilical cord blood serum (prepared in-house)). The medium was preheated for an hour in an incubator under 5% CO2 at 37 °C. Embryos were cultured statically under 5% CO2 at 37 °C. The total volume of the medium was replaced every 24 h, and the embryos were monitored by morphological assessment daily.
BMP inhibition in ex utero and explant cultures
For ex utero experiments, embryos were meticulously dissected at either E5.5 or E6.5 and processed for ex utero culture as described above. During the 24 h culture period, the embryos were co-cultured with 400 ng ml−1 of mouse recombinant Noggin (R&D systems, 1967-NG). Continuous morphological monitoring was conducted, and comprehensive analysis was performed using multiplexed RNA in situ HCR, as described below. In ExE explant cultures, embryos at E6.5 and 7.5 were dissected, and the ExE and EPC were precisely isolated from the embryonic compartment. Subsequent culture procedures followed an established protocol41, with the addition of 800 ng ml−1 of mouse recombinant Noggin or 1000 ng ml−1 mouse recombinant BMP4. After incubation for 24 h, bulk RNA was extracted and purified using the Micro RNA kit (Qiagen, 74004). qPCR was then used to quantify selected markers (primer details are provided in Supplementary Table 4). For temporal depletion of BMP signalling, embryos were dissected at E7.5 and were subjected to ex utero culture as described above. During the 12/24 h culture period, the embryos were co-cultured with 5 µM LDN-193189 (Sigma-Aldrich, SML0559), followed by whole-mount immunostaining.
Immunostaining
In this study, whole-mount immunostaining was performed as previously described24, using the following antibodies: rabbit anti-EOMES (1:100, ab23345, Abcam); rabbit monoclonal anti-Brachyury (D2Z3J) (1:100, Cell Signaling, 81694); mouse anti-KRT7 (1:100, Abcam; ab9021); goat anti-SOX2 (1:100, R&D, AF2018); rabbit anti-TFAP2C (1:100, CST, 2320); donkey anti-rabbit IgG (IgG) (H+L), Alexa 647 (1:250, Jackson ImmunoResearch, 711-605-152); donkey anti-goat IgG (H+L) Alexa 488 (1:250, Jackson ImmunoResearch, 705-545-003); goat anti-mouse IgG1 Alexa Fluor 594 (1:250, Jackson ImmunoResearch, 115-585-205).
Spatial analysis
Multiplex RNA in situ HCR22 was performed according to the manufacturer’s instructions (Molecular Technologies). For sample preparation, embryos were dissected and their Reichart’s membrane removed in M2 medium at consecutive timepoints after implantation until E7.5. Embryos were then washed in cold PBS and fixed in 4% PFA 4 °C overnight. After fixation, the samples were dehydrated on ice in increasing ratios of methanol (Sigma-Aldrich) and PBST (0.1% Tween-20) (Sigma-Aldrich) until freezing overnight in absolute methanol −20 °C. The embryos were then rehydrated in increasing ratios of PBST and methanol on ice, washed in PBST, treated with 10 μg ml−1 proteinase K (Thermo Fisher Scientific) and subjected to post-fixation in PFA 4% all at room temperature. For HCR staining, the samples were repeatedly washed with PBST, prehybridized with probe hybridization buffer (Molecular Technologies) for 30 min at 37 °C and then hybridized with probe sets (Molecular Technologies) for different combinations of Adm, Sox2, Eomes, Fgfr2, Ascl2, Bmp4, Lefty1, Fosl1, Chsy1, Hand1 and Pcdh12 (16 nM) at 37 °C overnight. Tissues were washed with HCR probe wash buffer (Molecular Technologies), followed by repeated washes in 5× SSCT (5× SSC with a final concentration of 0.1% Tween-20) and incubated with HCR amplifiers (Molecular Technologies) (30 pmol) in amplification buffer (Molecular Technologies) at room temperature overnight. The samples were then washed with 5× SSCT, labelled, mounted and imaged in an eight-well glass bottom/ibiTreat μ-plates (iBidi; 80827/80826). Spatial analysis was conducted using the Leica STELLARIS 8 Spectral confocal microscope and acquired using LAS-X (Leica). Fluorophores were excited by a white light laser and acousto-optical beam splitter. All embryonic specimens were visualized by maximum-intensity projection of their fluorescence signals across focal planes, and their 3D structure was assessed. The presented images were finalized using ImageJ software.
Flow cytometry
For isolation of single cells for scRNA-seq analysis, embryos were dissociated with 0.25% trypsin-A, 0.02% EDTA (Biological Industries) solution for 5 min at 37 °C and resuspended in DMEM without phenol red (GIBCO) supplemented with 10% FBS (Biological Industries). The samples were run on the FACS Aria-III flow cytometer (BD Biosciences, using BD FACSDiva v.9.0) using the ‘index sort’ option to retain the spectral properties of each individual sorted cell. For samples of ΔPE-Oct4-GFP44 obtained after E7.5, we further dissected areas on the basis of the localization of GFP expression. Subsequently, we used index-sorting and enriched for GFP-positive cells. The gating and sorting strategy is shown in Extended Data Fig. 13c. FlowJo v.10.7 was used to generate Extended Data Fig. 7c.
scRNA-seq analysis
10x Genomics
Cultured embryos were prepared for sequencing at different developmental stages after 2 days in culture: late streak and early head fold, assessed by light microscopy. Six similar embryos were selected for each developmental stage, pooled and dissociated using trypsin-EDTA solution A 0.25% (Biological Industries, 030501B) for 5 min at 37 °C. Trypsin was neutralized with medium that included 10% FBS and cells were washed and resuspended in 1× PBS (calcium and magnesium free) with 400 μg ml−1 BSA. Cell suspension was then filtered with a 70 μm cell strainer to avoid cell clumps. Single-cell viability was determined by trypan blue staining, before being diluted to a final concentration of 1,000 cells per μl. scRNA-seq libraries were generated for each pool of embryos separately using the 10x Genomics Chromium v3 system (5,000 cell target cell recovery) and sequenced on the Illumina NovaSeq 6000 platform according to the manufacturer’s instructions.
MARS-seq
Single-cell cDNA plate based libraries were prepared as previously reported68,69 according to the MARS-seq protocol70, following index FACS as described above.
scRNA-seq data processing
10x Genomics data from ex utero cultured embryos
Raw files were transformed into count matrices using Cell Ranger v.6.1.2 with the default parameters and with the prebuilt Cell Ranger reference package refdata-gex-mm10-2020-A (mm10 genome, GENCODE vM23/Ensemble 98). Cells with less than 2,000 counts, more than 30,000 counts or a high number of counts from mitochondrial genes relative to the number of counts from ribosomal genes were removed, resulting in 9,387 cells from the batch of late streak embryos and 3,916 cells from the batch of head fold stage embryos. For doublet removal, we ran DoubletFinder separately on the two batches, following the best practice workflow of the package. In brief, after creating a Seurat object for each batch69, Seurat principal component analysis was performed on the basis of the 2,000 most variable feature genes. For doublet detection using DoubletFinder, we calculated the fraction of artificial nearest-neighbour doublet cells (pANN) for each cell using pN = 0.25 (the relative frequency of artificial doublet cells relative to real cells) and pK = 0.02 for the batch of late streak embryos and pK = 0.01 for the batch of head fold embryos (pK is the relative neighbourhood size for the estimation of pANN). In the case of the late streak stage batch, we removed all cells with pANN > 0.2 (N = 1,753); for the batch of head fold stage embryos, we removed all cells with pANN > 0.25 (N = 291).
MARS-seq
MARS-seq libraries were sequenced using the NextSeq 500 or NovaSeq 6000 system. Reads were processed according to the MARS-seq2.0 protocol70 with the same specifications as previously reported12 using the STAR aligner for read alignment. Overall, we processed 129,024 wells, including the 40,868 wells of the previous version of the gastrulation atlas.
Metacell analysis and atlas construction
The basic idea of metacells is to partition cells into small groups of homogeneous cells, thereby removing the sparsity of RNA transcript counts associated with scRNA-seq technologies. Throughout the Article, we used metacell sizes of around 20–100 cells. For each metacell m, the absolute expression eg,m of a gene g is defined as the total number of transcript counts of this gene among all cells belonging to this metacell, normalized to the total number of counts, that is, \({e}_{g,m}={\sum }_{\{c\in m\}}{N}_{g,c}/({\sum }_{g}{\sum }_{\{c\in m\}}{N}_{g,c})\), where Ng,c is the number of the number of transcripts of gene g in the cell c.
Embryonic and extraembryonic WT atlas
To identify feature genes for metacell construction13, we selected all genes satisfying a minimal variance over mean (T_vm = 0.1) and coverage threshold (T_tot = 50 and T_top3 = 3). These 1,534 filtered genes were clustered into 120 clusters on the basis of their gene–gene correlation across the manifold. We manually selected and removed clusters enriched with cell-cycle- or stress-related genes, leaving 1,386 feature genes. The final metacell object (Knn = 100, minimal metacell size = 20) contained 983 metacells comprising 67,843 cells, including cells from E4.5 blastocysts.
Metacell object of ex utero culture embryos
The initial set of 1,017 feature genes was clustered into 60 groups and cell-cycle- and stress-related groups were removed. The metacell cover (148 metacells, 11,684 cells) was constructed on the basis of the remaining 829 feature genes and using the same parameters as above.
Metacell2 object of Bmp4-KO embryos
Using the framework of Metacell2 (ref. 14), all cells from the Bmp4-KO experiment were combined into a single-metacell object, including 6,705 cells from germline Bmp4-KO embryos, 11,968 cells from embryonic Bmp4-KO mutants, as well as 2,380 cells from control embryos. Metacells were constructed using the default parameters and a target size of 150,000 UMIs per metacell.
Embryo selection and temporal ordering
In total, cells from 287 individual embryos contributed to the embryonic and extraembryonic WT atlas. This includes cells from 153 embryos used in a previous version of the WT atlas12. Moreover, we collected 114 cells (after quality control) from two pooled samples of E4.5 blastocysts (n = 11 embryos). We selected 251 embryos with a sufficient number of embryonic cells for assigning them a developmental time (shown in Fig. 1c). The frequencies of ExE cell types were estimated on the basis of a cohort of 83 embryos (Extended Data Fig. 2b–d). This cohort included all of the embryos for which we collected the whole ExE tissue during dissection, for which we have a bright-field image to assign them a morphological stage and for which we obtained at least 10 ExE cells after quality control and filtering. Embryos with a sufficient number of embryonic cells (251 embryos) were temporally ordered as previously reported12. In brief, using the embryonic cells of an embryo, we calculate an embryo-embryo similarity matrix (Extended Data Fig. 1e) that quantifies the transcriptional similarity between two embryos. Using the similarity matrix, we compute a global goal function for each possible embryo order. Embryos are initially ordered on the basis of their morphology and then are reshuffled; each reshuffling is accepted if it improves the goal function. The final temporal ranks of each embryo are translated into developmental times (denoted as Et) by a spline interpolation of the nominal time of collection of each embryo versus its inferred transcriptional rank.
Network flow model construction
In a previous publication12 we introduced a network flow model that enabled us to reconstruct cellular differentiation trajectories during mouse gastrulation along the single-embryo single-cell transcriptome atlas. To infer cellular trajectories on the extended gastrulation atlas, temporally ranked embryos were grouped into 16 time bins (Fig. 1c). The network flow model for all embryonic cells was computed as previously reported. Logistic distances between metacells were calculated using the default parameters. For the estimated proliferation rate of each metacell, we interpolated between the default rate (3.5 divisions per day) and no cell division on the basis of each cell’s expression of cell-cycle-related genes. We used the same values for all additional network flow parameter as in the in the original network flow model of mouse gastrulation.
Differential expression statistical analysis
The expression of a gene between two groups of cells is compared using a χ2 test on the number of UMIs from that gene. If \({N}_{g}^{1}\) and \({N}_{g}^{2}\) are the number of UMIs per gene g and N1, N2 the total number of UMIs per group, we compute for each gene the χ2 statistic between the two two-dimensional vectors \(({N}_{g}^{1},{N}^{1}-{N}_{g}^{1})\) and \(\left({N}_{g}^{2},{N}^{2}-{N}_{g}^{2}\right)\). If multiple hypotheses are tested, P values are corrected using the Benjamini–Hochberg method.
Cell cycle scores
For each cell, its synthesis phase (S phase) and mitosis phase (M phase) score is the total number of UMIs from a list of respective marker genes, divided by the total number of UMIs of the cell. The M phase marker genes are as follows: Mki67, Cenpf, Top2a, Smc4, Ube2c, Ccnb1, Cdk1, Arl6ip1, Ankrd11, Hmmr, Cenpa, Tpx2, Aurka, AB349069, Kif4, Kif2c, Bub1b, Ccna2, Kif23, Kif20a, Sgol2a, Smc2, Kif11, Cdca2, Incenp and Cenpe. The S phase marker genes are as follows: Pcna, Rrm2, Mcm5, Mcm6, Mcm4, Ung, Mcm7, Mcm2, Uhrf1, Orc6 and Tipin.
Single-cell scores
Given a list of genes (referred to as cell-state markers) Gm, the single-cell score corresponding to a particular cell is calculated as the sum of counts for all genes in a given cell-state markers Gm, divided by the total sum of expression counts for all genes in the cell.
EPC lineage analysis
Let eg,m be the absolute expression for each gene g and all metacells m from the EPC lineage (uncommitted EPC, TGC progenitor, SpT-Gly), normalized to the total number of counts per metacell, and let leg,m = log2(eg,m + 10−5). We selected all variable genes that (1) pass a threshold of minimal expression in at least one of the metacells, that is:
$$\mathop{\min }\limits_{m}l{e}_{g,m}\, > \,-13$$
and that (2) pass a threshold on the difference between the highest and smallest expression in a metacell:
$$\mathop{\max }\limits_{m}l{e}_{g,m}-\mathop{\min }\limits_{m}l{e}_{g,m} > 2.$$
A complete list of these genes is provided in Supplementary Table 1. For further clean-up, we filtered only genes that show a minimal difference in their maximal expression among TGC progenitors and SpT-Gly metacells:
$$|\mathop{\max }\limits_{m\in \text{TGC}}l{e}_{{gm}}-\mathop{\max }\limits_{m\in \text{SpT}-\text{Gly}}l{e}_{{gm}}| > 1.5.$$
We observed that the large majority of the filtered genes followed one of the following behaviours: (1) high expression in TGC progenitor metacells compared to SpT-Gly and vice versa; (2) high expression in early metacells and low expression in late; or (3) low expression in early metacells and high expression in late ones. The filtered genes were therefore grouped into five clusters (arguing that this number should be enough to capture the above behaviours; Extended Data Fig. 3a) using k-means on the relative expression profiles, \(l{f}_{g,m}\,=l{e}_{g,m}-{\text{mean}}_{m}l{e}_{g,m}\). Genes from clusters 1 and 2 were used for the SpT-Gly score and genes from the clusters 4 and 5 were used for the TGC progenitor score. For pseudotime kinetics of gene expression, we fitted a principal curve to the joint distribution of Uncommitted EPC, SpT-Gly and TGC progenitor scores for all cells from the EPC lineage (Extended Data Fig. 3b) and divided the curve into 12 bins (Fig. 2a,b).
Chorion lineage analysis
Variable genes were filtered for all metacells from the chorion lineage (chorion progenitors, chorion) using the same parameters as for the EPC lineage (Supplementary Table 3). As in the EPC lineage, we also only filtered genes with
$$|\mathop{\max }\limits_{m\in \text{Chorion prog.}}l{e}_{{gm}}-\mathop{\max }\limits_{m\in \text{Chorion}}l{e}_{{gm}}| > 1$$
and clustered their relative expression into 5 clusters (Extended Data Fig. 4a). Genes from clusters 1 and 2 were used for chorion progenitor score and genes from clusters 4 and 5 for the chorion score. Gene expression kinetics (Fig. 2c,d) is shown along the principle curve fitted to the joint distribution of chorion progenitor and chorion scores for each chorion and chorion progenitor cell along 12 bins (Extended Data Fig. 4b).
Cell type annotation of cells from embryos with genetic manipulations
Cells from EXE Elf5-KO, all Bmp4-KO models and control embryos were annotated with a cell type from the WT atlas as previously described14,29. For each experiment, we constructed a joint metacell k-nearest neighbours similarity graph consisting of query cells from the KO embryos and WT atlas cells. For each query and each atlas cell, we sampled the empirical distribution of cell types from atlas cells among its 100 nearest neighbours. Each query cell is matched with an atlas cell (and annotated with its cell type) on the basis of matching the empirical cell type distribution of the query cell with the best-correlated cell type distribution among the atlas cells. Query cells with less than ten atlas cells among their nearest neighbours were assigned with an atlas cell type by computing the cell-metacell correlation corg(log2(ug,c + 1), log2(eg,m + 10−5)) for each atlas metacell over the list of feature genes and using the cell type of the best-correlated atlas metacell.
Developmental timing of Elf5-KO and Bmp4-KO embryos
Given a temporal order of embryos from the WT atlas, query embryos were assigned a best-matching WT rank as previously reported14,29: using the joint metacell k-nearest neighbours similarity graph of query cells and atlas cells, each query cell is annotated with the temporal rank of the nearest neighbour atlas cell (that is, the temporal rank of the embryo this cell belongs to). In a similar way, we resample for each atlas cell a temporal rank by assigning to it the rank of the nearest neighbour cell from a different atlas embryo. As a result, we obtain for each embryo a distribution of sampled temporal ranks of the cells belonging to the embryo. Each query embryo is mapped onto a WT rank by computing the correlation between the cumulative distributions over ranks between the query embryo and the WT embryos and using the temporal rank of best-correlated WT embryo.
Atlas projection and cell type annotation of cells from ex utero cultured embryos
Each cell from ex-utero-cultured embryos was projected onto the WT atlas by matching its expression profile with the best-matching atlas metacell profile: Let ug,c be the single-cell gene expression matrix for all cells c> from ex utero cultured embryos and eg,m the WT atlas gene expression per metacell m for all genes g from a set of 581 feature genes. On the basis of the cell-metacell correlation matrix
$${\text{cor}}_{g}({\log }_{2}{(u}_{g,c}+1),{\log }_{2}({e}_{g,m}+{10}^{-5}))$$
each cell is matched with its best-correlated metacell and annotated with the cell type of the metacell.
Similarly, each metacell from ex utero cultured embryos is matched with its best-correlated WT atlas metacell using the correlation matrix
$$\text{cor}({\log }_{2}{(u}_{g,k}+1),{\log }_{2}({e}_{g,m}+{10}^{-5}))$$
where ug,k is the metacell expression matrix per gene g and metacell k from the ex utero cultured embryos.
Cell type frequency comparison between ex utero cultured and WT embryos
To compare the distribution of cell states in ex utero cultured embryos to the WT distribution, we first computed for each batch of ex utero cultured embryos (late streak and late head fold) a best-matching developmental time, thereby correcting for potential differences in the mean developmental time of the two groups. Ex utero cultured embryos were assigned an average developmental time using for each ex utero cell c the best-matching atlas metacell m(c). For a batch b of ex utero embryos, let \({p}_{m}^{b}\) be the number of ex utero cells for which m(c) = m normalized to the total number of cells, that is, the number of cells that project to that atlas metacell normalized by the total number of cells from that batch. Vice versa, for each WT embryo e (indexed here by their temporal rank 1, 2, …, 235), let \({p}_{m}^{e}\) be the number of cells from that embryo in the metacell m normalized to the total number of cells from that embryo. As the number of cells per metacell is low for single embryos, we use instead of \({p}_{m}^{e}\) the averaged frequency of cells \({\bar{p}}_{m}^{e}={\sum }_{f,|f-e|\le w}{p}_{m}^{f}\) for comparison with \({p}_{m}^{b}\) (over a time window w). More precisely, each ex utero batch was matched with that WT embryo e for which thedistance between the two distributions \({p}_{m}^{b}\) and \({\bar{p}}_{m}^{e}\) was minimal, that is,
$$\text{matched WT rank}\,=\,{{\rm{argmin}}}_{e}\,|{p}_{m}^{b}-{\bar{p}}_{m}^{e}|.$$
For the batch of late streak embryos we used a window size w = 20 and for the batch of late head fold embryos window size w = 5.
Differential expression analysis for Bmp4-KO embryos
Identification of batch-related genes and metacell–metacell projection
We created a metacell object of all cells from the Bmp4 experiment. Metacells from the Bmp4 experiment were projected onto the WT atlas using the framework of MCProj14. This approach enables us to match each query metacell m and its expression profile \({e}_{g,m}^{{\rm{query}}}\,\) (g is a gene) with a corresponding matched WT atlas profile \({e}_{g,m}^{{\rm{proj}}}\). Both profiles represent the relative absolute expression, that is, \(1={\sum }_{g}{e}_{g,m}^{{\rm{query}}}={\sum }_{g}{e}_{g,m}^{{\rm{proj}}}\). To screen for batch-related genes, we computed the log fold changes between pseudo-bulk query and atlas expression profiles for all cells c from a cell typet and experimental condition b, that is:
$${lfc}_{g,t}^{b}={\log }_{2}\left(\frac{1}{{N}_{b,t}}\sum _{c\in (b,t)}{e}_{g,m(c)}^{{\rm{query}}}+\varepsilon \right)-{\log }_{2}\left(\frac{1}{{N}_{b,t}}\sum _{c\in (b,t)}{e}_{g,m(c)}^{{\rm{proj}}}+\varepsilon \right)$$
where m(c) is the metacell m of the cell c, Nb,t is the number of cells from cell type t and experimental condition b and ε = 5 × 10−5. There are four different experimental conditions: (1) homozygous Bmp4Δ/Δ cells from germline Bmp4Δ/+ mating (characterized on the basis of total low levels of BMP4 per embryo), (2) heterozygous Bmp4Δ/+ or Bmp4+/+ cells from germline Bmp4Δ/+ mating (normal levels of BMP4), (3) cells from embryonic Bmp4-KO mutants (tetraploid) and (4) isogenic control cells from injected (tetraploid) WT embryos (Extended Data Fig. 11a,b). We subsequently clustered all differentially expressed genes (genes g for which \(\mathop{\max }\limits_{t,b}|{{lfc}}_{g,t}^{b}| > 0.8\) and that pass a minimal expression threshold) into ten clusters. Genes from clusters displaying differential expression among the control cells were classified as lateral genes.
Differential expression per embryo and cell type in the ExM lineage
For each query embryo e, we computed the bulk expression per cell type t and gene g, \({f}_{g,e,t}^{q}\) and the bulk expression per cell type of time-matched WT embryos \({e}_{g,e,t}^{{\rm{WT}}}\). To analyse the effect of embryonic Bmp4-KO on the ExM lineage, we selected all of the expression profiles from the cell types early nascent mesoderm, ExM and allantois and from the control and embryonic Bmp4-KO mutants that contained at least ten cells per cell type and embryo. We then filtered all of the genes for which (1) \(\mathop{\max }\limits_{e,t}| {\log }_{2}(\,{f}_{g,e,t}^{q}+\varepsilon )\,-{\log }_{2}(\,{f}_{g,e,t}^{{\rm{WT}}}+\varepsilon )| \ge {\log }_{2}3\), that is, displaying a threefold change between embryonic Bmp4-KO and matched WT cells in at least one of the cell types and one of the embryos; and (2) passing a threshold on minimal expression in one of the conditions, that is, \(\mathop{\max }\limits_{e,t}(\,{f}_{g,e,t}^{q},{f}_{g,e,t}^{{\rm{WT}}}\,)\ge 1\times 1{0}^{-4}\). After removing lateral genes (see the previous paragraph), this resulted in 46 genes, which were subsequently clustered into three groups. Cluster 2 is shown in Extended Data Fig. 12c. Genes of clusters 1 and 3 are listed in Supplementary Table 5.
Identification and differential gene expression of ExM PGC precursors
Let eg,m be the absolute expression for each gene g and all metacells m from the PGCs, allantois and ExM, normalized to the total number of counts per metacell, and let leg,m = log2(eg,m + 10−5). We selected all variable genes that (1) pass a threshold of minimal expression in at least one of the metacells, that is:
$$\mathop{\min }\limits_{m}l{e}_{g,m}\, > \,-13$$
and that (2) pass a threshold on the difference between the highest and smallest expression in a metacell:
$$\mathop{\max }\limits_{m}l{e}_{g,m}-\mathop{\min }\limits_{m}l{e}_{g,m} > 4.$$
A complete list of these genes is provided in Supplementary Tables 6 and 7. For further clean-up, we filtered only genes that show a minimal difference in their maximal expression among PGCs and Allantois or ExM metacells:
$$|\mathop{\max }\limits_{m\in \text{PGC}}l{e}_{{gm}}\,-\,\mathop{\max }\limits_{m\in \text{Allantois},\text{ExM}}l{e}_{{gm}}\,| > 1.5.$$
As in the ectoplacental cone lineage, the filtered genes were therefore grouped into five clusters using k-means on the relative expression profiles, lfg,m = leg,m − meanmleg,m. Genes from clusters 1 were used to calculate the PGC score, and genes from clusters 3, 4 and 5 were used to calculate the allantois–ExM score. On the basis of these scores, we identified a population of 10 metacells consisting of 164 cells that are probably PGC precursors. These metacells were labelled as ExM PGC precursors on the basis of their intermediate-level PGC score and their early developmental time (Fig. 5i–k). Of the 164 cells, 32% originated from embryonic Bmp4-KO mutants, 52% from ΔPE-Oct4-GFP embryos (that is, PGC enrichment assay, see above) and the remaining 16% were from the atlas. To investigate differential gene expression, we compared PGC precursor cells from embryonic Bmp4-KO mutants with those from WT embryos (Fig. 5k) as described above. We also compared PGC cells from embryonic Bmp4-KO mutants to matched WT cells, which were a subset of cells from each group that was stratified by its PGC score (Extended Data Fig. 12d).
Estimated and sampled number of EXE cells per age group
The representativeness of cells in the ExE was rigorously validated using a dual-pronged approach. Initially, the ExE-to-embryonic cell ratio was computed for each age group, offering a quantitative assessment of the relative abundance of ExE cells throughout the developmental stages. Complementing the ratio calculation, nucleus count data from a previous study12,14 were used to estimate ExE cell numbers across diverse developmental stages. The datasets were harmonized by systematically pairing each embryo in the current study with a corresponding embryo from the previous dataset, aligning them on the basis of morphological stages. The integrated datasets enabled a thorough analysis of the congruence between the sampled ExE cells in the current study and the anticipated cell numbers at each timepoint. This comparison, visually depicted in Extended Data Fig. 2d, highlights the close correspondence observed between the sampled and expected ExE cell numbers, affirming the robustness of the used cell sampling methodology.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.